 |
A 19-year-old male initially presented to the emergency eye clinic complaining of progressive blurred vision in the right eye that started three months earlier. He also reported a long history of decreased night vision affecting both eyes. The patient was not taking medication. His social history is noncontributory.
Upon examination, his visual acuity was measured to be 20/400 OD and 20/80 OS with no improvement after refraction. Extraocular motilities showed full range of motion. His confrontation fields were full-to-finger count in each eye. Pupils were round and reactive to light with no afferent defect. His color vision with the Ishihara plates was reduced to 5/9 plates OD and 6/9 plates OS. His intraocular pressures (IOP) were 16mm Hg OD and 17mm Hg OS.
Anterior segment examination yielded normal limits. In addition to an OCT of the retina (Figures 1a and 1b), we obtained fundus images (Figures 2a and 2b) that displayed optic nerves that were normal in appearance, but showed the presence of white pigmentary changes of the retinal pigment epithelium (RPE) along the arcades and macular changes (Figures 3a and 3b). Intravenous fluorescein angiography (FA) (Figures 4a and 4b) were also obtained.
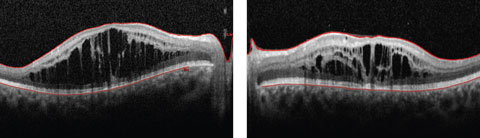 |
| Figs. 1a and 2b. This 19-year-old patient presented with progressive blurring in the right eye and decreased night vision. What can these OCT findings tell you? Click image to enlarge. |
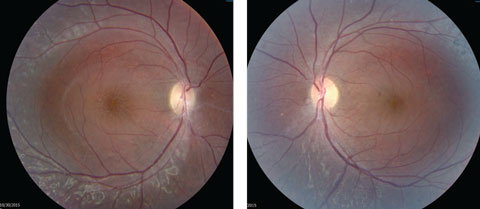 |
| Figs. 2a and 2b. Our patient’s optic discs appeared normal, but do these fundus images point to the cause of his issue? Click image to enlarge. |
Take the Quiz
1. What does the OCT reveal?
a. Vitreoretinal traction.
b. Retinal thickening with cystoid changes of the inner retina.
c. Retinoschisis of the nerve fiber layer.
d. All of the above.
2. What does the intravenous fluorescein angiography show?
a. Significant leakage of fluorescein in both the eyes.
b. Significant pooling of fluorescein in one of the eyes.
c. Areas of hypofluorescence that correlate with blockage of the retina from blood.
d. No significant leakage.
3. What is the likely diagnosis?
a. Enhanced S-cone syndrome/Goldmann-Favre Syndrome.
b. Retinitis pigmentosa.
c. North Carolina macular dystrophy.
d. Choroideremia.
4. What additional test would be most helpful to confirm the diagnosis?
a. Fine needle biopsy.
b. Fundus autofluorescence.
c. ERG/genetic testing.
d. B-scan ultrasonography.
5. What are some possible treatment options for this patient?
a. Oral or topical carbonic anhydrase inhibitor.
b. Photodynamic therapy.
c. Radiation.
d. Vitrectomy/membrane peel.
Discussion
The OCT of the retina showed profound macular cystoid changes with no leakage on the FA. This supported the conclusion that a non-leaking cystoid macular edema or a macular retinoschisis was present in the patient. In addition, the ERG showed reduced cone response and near absent rod response with increased cone responses under a blue stimulus. In following with symptoms of night blindness, reduced color vision and annular white pigmentary changes of the RPE along the vessels, the patient was suspected to have enhanced S-cone syndrome (ESCS).
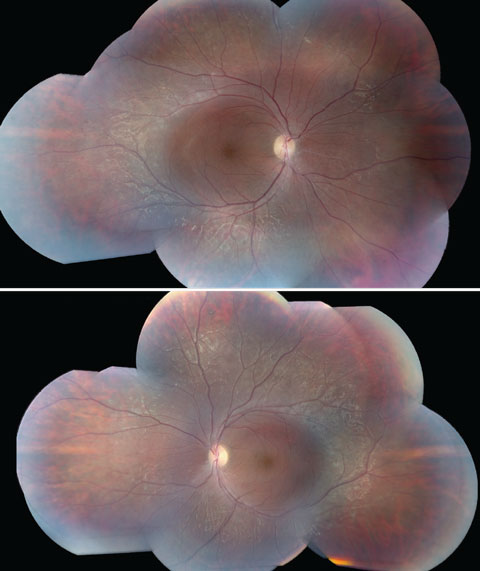 |
| Figs. 3a and 3b. These fundus images of the left (above) and right eyes show white pigmentary changes of the RPE along the arcades as well as macular changes. Click image to enlarge. |
Researchers postulate that enhanced S-cone syndrome and Goldmann-Favre syndrome (ESCS/GFS) fall under a spectrum of a single vitreoretinal degeneration.1 The inheritance pattern is autosomal recessive via a mutation in nuclear receptor gene NR2E3. The NR2E3 mutation encodes for a ligand-dependent transcription factor that is expressed in the photoreceptors and is involved in suppressing the expression of certain genes of rods and cones, and suppressing cone differentiation in early embryogenesis.1 The resulting mutation then causes an increased number of S-cones at the expense of other photoreceptors, and also may create a weakness in cellular adhesion, according to research speculation.2 The abundance of S-cones causes the characteristic increased sensitivity to short wavelengths during ERG testing, while the weakness in cellular adhesion is related to the development of cystoid macular changes, researchers believe.2
ESCS, along with its characteristic response to short wavelengths, typically shows a profoundly abnormal ERG with an absence of rod-driven response and can lead to severe depression of both the cone and rod response.3 In cases when the responses are extinguished and suspicion of the diagnosis still remains, genetic testing may provide further insight in supporting the diagnosis. In our patient’s case, following the conventional full-field ERG, the patient underwent an additional ERG test known as the S-cone protocol (Figure 5). This protocol involves saturating the L- and M- cones, such that only the functions of S-cones are tested. Under certain specific intensities, our patient exhibited a response with an increased amplitude of approximately two to three fold outside the normative values, supporting the evidence of S-cone sensitivity and our initial diagnosis.
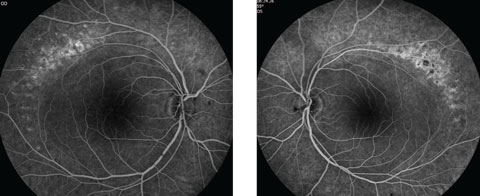 |
| Figs. 4a and 4b. Do our patient’s intravenous FA images reveal anything about his likely diagnosis? Click image to enlarge. |
The most commonly reported symptoms of ESCS include night blindness and progressive visual acuity loss.4 Clinical findings of ESCS/GFS are atypical pigmentation degeneration of the RPE in the mid periphery that may be accompanied by dendritic whitish retinal vessels, vitreous humor degeneration such as the appearance of an optically empty vitreous, and the presence of vitreous veils, foveal and peripheral retinoschisis, posterior subcapsular cataract and cystoid macular edema. The distinction between GFS from ESCS is that the former falls on the severe end of the wide disease spectrum.4
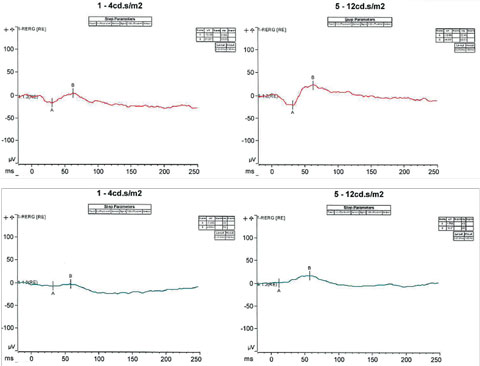 |
| Fig. 5. S-cone protocol testing involves saturating the L and M cones, such that only the functions of S-cones are tested. Under certain specific intensities, our patient exhibited a response with an increased amplitude of approximately two to three fold outside the normative values. Click image to enlarge. |
The use of oral and topical acetazolamide has long been proposed as a possible treatment of macular edema secondary to hereditary retinal dystrophies.2 The clinical effect of a carbonic anhydrase inhibitor is said to act on the membrane receptors of the RPE, where it may acidify the subretinal space, decrease its volume as well as help stabilize cellular adhesion between the retina and the RPE.2 However, this theory remains in question, given cases of ineffective outcomes of both topical and oral carbonic anhydrase inhibitors having rather ineffective outcomes.5 Other proposed treatments that have been investigated in the treatment of macular edema and retinoschisis include grid laser photocoagulation, cyclosporine A and observation.6-8 A general consensus on primary treatment still remains to be established.
Following the examination, the patient subsequently had genetic testing and had a positive finding for a heterozygous mutation in the NR2E3 gene. In addition, the patient was also started on 500mg of oral acetazolamide daily. At the patient’s two-month follow-up visit, the OCT displayed an improvement in the cystoid macular edema in each eye, with improved visual acuities of 20/30 OD and 20/50 OS.
Dr. Kim practices at Nova Southeastern University in Ft. Lauderdale, where he is also a faculty instructor.
|
1. Yzer S, Barbazetto I, Allikmets R, et al. Expanded clinical spectrum of enhanced S-cone syndrome. JAMA Ophthalmol. 2013 Oct;131(10):1324-30. 2. Salvatore S, Fishman G, Genead M. Treatment of cystic macular lesions in hereditary retinal dystrophies. Surv Ophthalmol. 2013 Nov-Dec;58(6):560-84. 3. Pachydaki S, Bhatnagar P, Barbazetto I, et al. Long-term follow-up in enhanced s-cone syndrome. Retina Cases Brief Rep. 2009;3(2):118-20. 4. Herrador-Montiel Á, Sánchez-Vicente J, Arias-Alcalá M. Clinical features of Goldmann-Favre vitreoretinal degeneration. Arch Soc Esp Oftalmol. 2012 Aug;87(8):260-2 5. BušićM, Bjeloš M, Bosnar D, et al. Cystoid macular lesions are resistant to topical dorzolamide treatment in enhanced S-cone syndrome child. Doc Ophthalmol. 2016 Feb;132(1):67-73. 6. Hayashi T, Gekka T, Tsuneoka H. Spontaneous resolution of large macular retinoschisis in enhanced s-cone syndrome. Ophthalmic Surg Lasers Imaging Retina. 2016 Feb;47(2):187-90. 7. Garweg J, Böhnke M, Mangold I. Treatment of Goldmann-Favre syndrome with cyclosporin A and bromocriptine. Klin Monbl Augenheilkd. 1991 Sep;199(3):199-205 8. Khairallah M, Ladjimi A, Ben Yahia S, et al. Elevated macular retinoschisis associated with Goldmann-Favre syndrome successfully treated with grid laser photocoagulation. Retina. 2002 Apr;22(2):234-7. |
