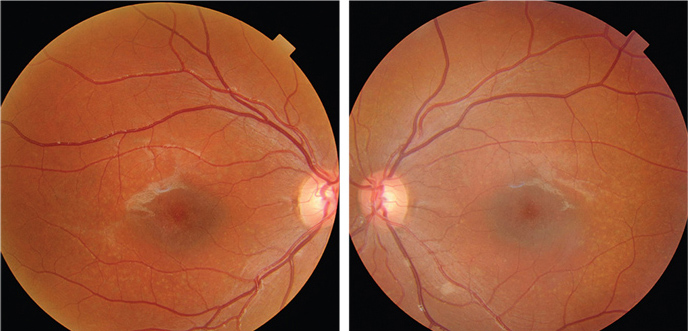
 |
| Fundus photographs of the eight-year-old son who presented with a visual acuity of
20/200 OU (OD left, OS right). |
Stargardt disease––with or without the deep white/yellow retinal lesions, or the so-called flecks of fundus flavimaculatus––is an inherited autosomal recessive trait caused by mutations in the ABCA4 gene.2,3
Symptoms usually begin in the first two decades of life, and final visual acuity generally stabilizes between 20/200 and 20/400.4
Both spectral domain optical coherence tomography (SD-OCT) and fundus autofluorescence (FAF) permit clinicians to detect and potentially manage Stargardt disease earlier than ever before. Using these imaging modalities, I’ve documented what I believe to be the earliest ocular abnormality associated with Stargardt disease––a thickened, irregular, hyper-reflective external limiting membrane (ELM).
These findings are described in the following case series.
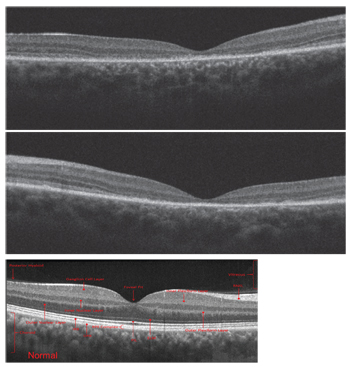 |
| Spectral-domain optical coherence tomography scans of the son
(OD top, OS middle), compared to a normal scan in a healthy individual (bottom). |
Upon referral, a mother presented with her three children: an eight-year-old son, a five-year-old daughter and a four-year-old daughter. Although her daughters had no visual symptoms and were never evaluated, her eight-year-old son complained of progressive, bilateral visual acuity reduction at distance and near over a three-year duration. He also complained of some difficulty with both color and night vision.
The mother reported that although 15 ODs and MDs examined her son over this period, no practitioner offered any organic explanation of his visual complaints. (She reported that several clinicians hinted at an underlying psychogenic cause.)
Careful questioning failed to reveal any family history of eye problems or contributory systemic disease.
Case I
The eight-year-old son’s best-corrected visual acuity measured 20/200 OU. We performed funduscopy and SD-OCT OU.
On SD-OCT, we ob-served macular thinning in his right eye. Also, we noted a general absence of the photoreceptor integrity line (PIL). It appeared that the son’s outer retina was collapsing onto the retinal pigment epithelium (RPE) in the entire central zone.
The PIL was somewhat preserved outside the macula; however, it should be present in the entire scan. We uncovered very similar SD-OCT findings in the son’s left eye.
Both the FAF images and the SD-OCT scans reveal marked abnormalities in the outer retinas of both eyes. Hyper- and hypo-FAF findings suggested a primary abnormality of the RPE, with secondary involvement of the overlying photoreceptors.
Flash ERG results were low/normal in amplitude, and not delayed under photopic and scotopic conditions OU. Such normal or near-normal ERG results effectively ruled out an overall cone-rod dystrophy.
We diagnosed the son with probable Stargardt disease and fundus flavimaculatus.
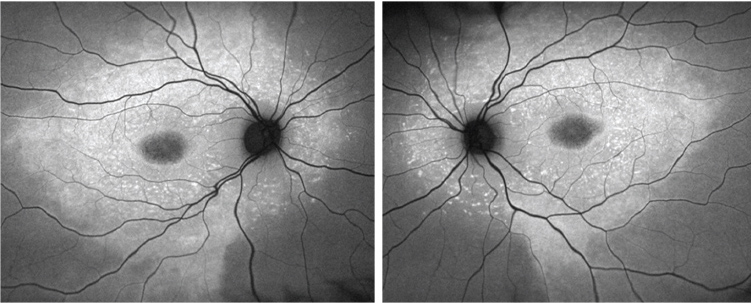 |
| Fundus autofluorescence images of the son (OD left, OS right). |
The five-year-old daughter exhibited no definitive symptoms of Stargardt disease. Her best-corrected visual acuity measured 20/25 OU, which is considered normal in four- and five-year-olds. The fundus images revealed normal, white, glistening reflections off the ILM in both eyes––an expected finding in young children. So, we interpreted the fundus evaluation as essentially normal OU. However, FAF testing revealed a subtle bull’s eye in both maculas.
In contrast to her brother, the five-year-old sister exhibited normal, intact retinal layering––although the PIL was somewhat attenuated. Further, the ELM was thickened, irregular and grossly hyper-reflective. In healthy individuals, the ELM appears very subtle and perhaps one-fifth to one-tenth as thick and reflective as the PIL. In both eyes, her ELM was thicker and more reflective than the PIL. Specifically, the abnormal ELM was most markedly evident in the macula.
Case III
The four-year-old daughter’s best-corrected visual acuity also measured 20/25 OU. Like her older sister, we documented characteristic ILM reflections on the fundus evaluation of both eyes.
With parental consent, we used chocolate to bribe the girl and improve her cooperation for the SD-OCT scan. Her images were very similar to those obtained of the older sister––her PIL was somewhat thinned, and the ELM was thickened, irregular and hyper-reflective OU.
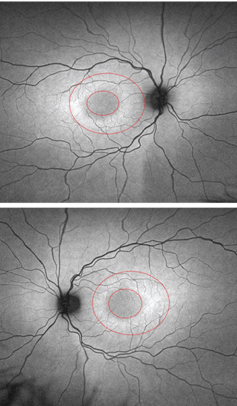 |
| FAF images of the five-year-old daughter
(OD top, OS bottom). Note the bull’s eye around both maculae. |
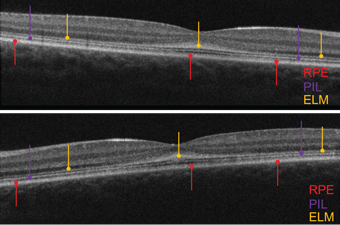 |
| OCT scans of the five-year-old daughter (OD top, OS bottom).
|
In comparison to their brother––who exhibited a thin macula and compromised outer retinal structures––the two sisters had intact retinal layers, but an abnormally thickened ELM. The apparent irregular thickening and hyper-reflective appearance on the OCT potentially was due to adjacent layer degeneration, with cellular debris accumulating on the ELM.5 The ELM––likely formed by the footplates of the Müller cells––is structural in nature and supportive of the retina’s neural components.
In the two sisters, the apparent ELM thickening likely was due to the close proximity of cellular debris deposits. The outer nuclear layer (ONL) normally is comprised of dense, closely packed nuclei of cones and rods, and hence is hyporeflective.
As the nuclei degenerate, they are less densely packed and occupy a larger space. But as this occurs, the entire ONL becomes less hyporeflective and more hyper-reflective. This newly hyper-reflective zone appears as an extension of the hyper-reflective ELM. This hypothesis is supported by some rare histopathological specimens.6
After a thorough evaluation and comparison of these three siblings, it seemed increasingly evident that the abnormal ELM appearance might be the earliest clinical indication of Stargardt disease.
To date, we have not observed a similar finding in retinitis pigmentosa or any other retinal degenerations. Indeed, this apparent thickened, irregular and hyper-reflective ELM may be a biomarker for very early Stargardt disease––even before the onset of symptoms and acuity reduction.
Another potential explanation for associated ELM thickening is a defect in the ABCA4 gene. In patients with Stargardt disease, a compromised ABCA4 gene allows toxic vitamin A dimers to accumulate in the photoreceptors.7
 Follow-Up
Follow-Up
All three siblings were evaluated four months after their initial visit. FAF imaging revealed bilateral progression in the eight-year-old son. His preliminary genetic testing results revealed the presence of at least one ABCA4 gene abnormality, which has been associated with Stargardt disease.
After meeting with an executive from Alkeus Pharmaceuticals, the mother agreed to allow her three children to be enrolled in a study testing its oral, chemically modified vitamin A supplement, ALK-001. It is anticipated that FDA testing of ALK-001 will begin within this calendar year.
Interestingly, both parents had normal fundus evaluations and SD-OCT scans. Because Stargardt disease typically has an autosomal recessive inheritance pattern, both the mother and father most likely had one abnormal gene. Unfortunately, in this particular instance, all three children inherited the abnormal recessive gene from both parents.
Dr. Sherman is a distinguished teaching professor at State University of New York College of Optometry and the Schnurmacher Institute of Vision Research. He also practices at The Eye Institute and Laser Center, New York City.
Thanks to Arnold Sherman, OD, and Jennifer Lee for their contributions to this article.
To view additional images and an expanded discussion, go to www.retinarevealed.com and click on Case #55 in the archive.
1. Stargardt KB. Über familiäre, progressive Degeneration in der Makulagegend des Auges. Albrecht von Graefes Archiv für Ophthalmologie. 1909;71:534-50.
2. Lois N, Halfyard AS, Bird AC, et al. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol. 2004 Jul;138(1):55-63.
3. Ozdek S, Onaran Z, Gürelik G, et al. Stargardt’s disease and retinitis pigmentosa: different phenotypic presentations in the same family. Eye (Lond). 2005 Nov;19(11):1222-5.
4. Rotenstreich Y. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003 Jun;110(6):1151-8.
5. Burke TR, Yzer S, Zernant J, et al. Abnormality in the external limiting membrane in early Stargardt disease. Ophthalmic Genet. 2013 Mar-Jun;34(1-2):75-7.
6. Gregory-Evans K, Fariss RN, Possin DE, et al. Abnormal cone synapses in human cone-rod dystrophy. Ophthalmology. 1998 Dec;105(12):2306-12.
7. Alkeus Pharmaceuticals Inc. Preclinical Results: ALK-001 halts vision loss in a mouse model of Stargardt disease. Available at: www.alkeus.com/preclinical.html. Accessed June 3, 2013.
