The retinal dystrophies belong to a group of diseases sometimes referred to as orphan diseases. This term is reserved for medical conditions that affect fewer than 200,000 persons in the United States. The diagnosis of these dystrophies is usually based on patient symptoms, the clinical picture and test results. Some of these dystrophies are progressive and can lead to severe visual loss, and medical treatment is currently nonexistent. But, ongoing clinical trials will most certainly allow for medical options within the next decade.
We are standing on the edge of change. The Human Genome Project ushered in a whole new era in the discovery of genes and genetic mutations, which will help our understanding of the diagnosis, treatment and prevention of many genetic diseases. The project also spawned a new terminology: proteomics, the study of the proteins produced by specific genes and how these proteins affect structure and function.
Theres been an explosion in genomic information of the orphan retinal diseases in the past decade, with new mutations being identified almost monthly.
So, how do the genomics of the orphan retinal diseases affect your practice? And, how will these discoveries affect the way in which you diagnose and manage patients who have these retinal diseases? Currently, many practitioners are unable to identify a retinal dystrophy and usually refer such cases to a medical retinal specialist who is an expert in the retinal dystrophies. However, genetic tests are now available to confirm the diagnosis of some diseases. And, there are genetic specialists in many communities with whom you can comanage your patients, much as you comanage refractive surgery and cataract patients. In addition, these discoveries guide research leading to treatment modalities, which will have the potential to prevent retinal damage, rescue damaged cells and delay progression of disease.
This article is the first in a three-part series describing what we currently know about the genetic developments in some of the hereditary eye diseases and their impact on your practice. There are many retinal diseases. But due to space limitations, the epidemiology, symptoms, diagnostic tests and genetics of only the more commonly seen hereditary retinal diseases are discussed, so you will know what to do, should a patient present with a suspected hereditary disease.
Retinitis Pigmentosa
Retinitis pigmentosa (RP) is the most commonly seen hereditary retinal disease. It is actually a group of diseases that can have a common phenotypic presentation. RP can occur alone or as part of a syndrome, such as Ushers syndrome, in which hearing loss is associated with retinal degeneration. RP affects about one in 3,500 people worldwide.1 In the United States, approximately 100,000 people are affected.1
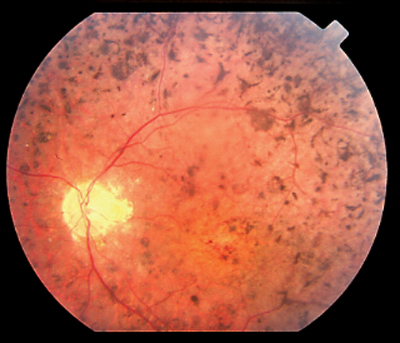 |
| Bone-spicule pigmentation and narrow arterioles are classic signs of retinitis pigmentosa. |
All forms of RP result in photoreceptor degeneration, to varying degrees. The classic symptoms of RP are night-blindness, which can occur at any age depending on the type of RP, and peripheral visual field loss. About half of RP cases not associated with a syndrome can occur without any family history, This makes the diagnosis more difficult.1
Of the other half of RP cases that do have a positive family history, 40% are either dominant or recessive, and another 10% are sex-linked (the disease is inherited on the X chromosome.)2 Symptoms usually occur later in life in the dominant form and are not as severe as with other types of RP. In recessive and sex-linked RP, symptoms typically occur in the early teens, and vision can be severely affected by middle age.
Clinical findings can also be classic, but sometimes they are not so clear. The most consistent finding in RP is arteriolar attenuation. Other findings can include (to varying degrees) bone-spicule pigmentation, caused by pigment proliferation along the blood vessels; a grayish retinal sheen; pale, waxy disc, sometimes with disc drusen; cystoid macular edema; and posterior subcapsular cataract. Inflammatory diseases can sometimes mimic these appearances and are often referred to as pseudo-RP.
Visual field testing will initially show a ring scotoma in the mid-periphery. This will spread peripherally and centrally resulting in tunnel vision visual fields.
One clinical pearl: If retinal changes are fairly symmetric between the eyes, you are likely dealing with a hereditary disease, especially in a young patient. And, this clinical pearl applies to all hereditary eye diseases, not just RP.
Phenotypic presentations of RP can vary. RP can occur without any pigmentary disturbances, called RP sine pigmento. It can also affect only a part of the retina as in sector RP. Or, RP can be accompanied by numerous punctate white spots. This is called retinitis punctata albescens.
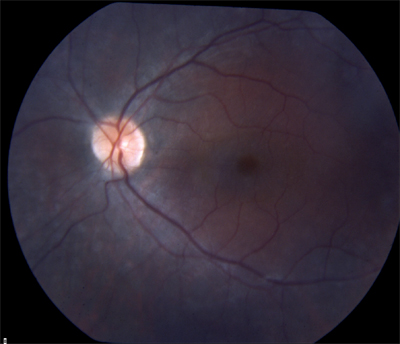 |
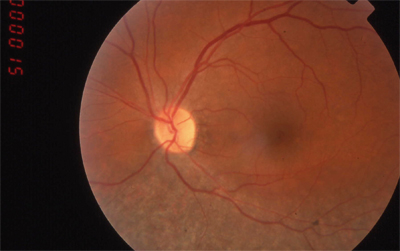 |
| RP sine pigmento (without pigment). | Sector RP affects only part of the retina. |
Diagnosis of RP is most often made based on the patients symptoms, the clinical picture, visual fields and specialized electrodiagnostic testing. Electrodiagnostic testing is offered at some private practices but is usually only available at special testing facilities associated with schools of optometry or in university medical centers.
Chief among these is the full-field electroretinogram (ERG). The ERG detects a retinal response to a flash of light that comes from the photoreceptors (which give rise to the a-wave) and the Mueller and bipolar cells of the inner retina (which give rise to the b-wave). The ERG, using a dim white light, is performed first on a dark-adapted retina to isolate rod response and then in a light-adapt-ed retina to isolate cone response. The ERG response in RP is significantly reduced in amplitude or completely flat under both light-adapted and dark-adapted conditions (some testing centers may use multifocal systems that can test ERG responses at many points on the retina).
In the United States, mutations in the RHO (rhodopsin) gene account for about 30% of dominant RP.3 Of the mutations seen in the RHO gene, about 12% to 15% are from a single mutation: Pro23His. Mutations in the peripherin/RDS gene account for 8% to 10% of dominant RP.3
Mutations in other genes that cause dominant RP (RP1, CRX, NRL, RGR, IMPDH1, PRPF3, PRPF8, PRPF31, RP9, CA4 and Fscn2) account for 15% to 20% of dominant RP, in which the genes have been identified.1 Despite what has been discovered, researchers have not yet identified the genes that cause the remaining 45% to 50% of dominant RP.
Recessive RP is caused by about 60 different genes. Sex-linked RP is a very severe form of RP caused by two known genes.4 About 75% of X-linked RP is caused by mutation in the RPGR gene. A second gene for X-linked RP, RP2, causes about 25% of cases.
Genes synthesize proteins and are named based on the protein they produce. (For a description of these genes, the proteins for which they are named and the diseases for which they are associated, see Genes Implicated in Non-Syndromic Retinitis Pigmentosa and Related Dystrophies, below.) These proteins are ultimately re-sponsible for the visual cycle that regenerates rhodopsin and transduces light energy into an electrochemical signal (below). Any missing or abnormal protein prevents these processes from occurring and therefore results in reduced vision.
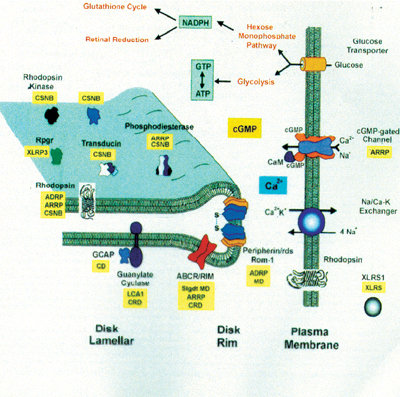 |
| The visual cycle requires specific proteins (depicted in black). Mutations in these proteins are associated with certain retinal dystrophies (depicted in yellow) (ADRP= autosomal dominant RP, ARRP= autosomal recessive |
Genes Implicated in Non-Syndromic Retinitis Pigmentosa and Related Dystrophies1
Group of Genes and Their Function
Gene
Protein Produced
RP or Related Retinal Dystrophy
I. Phototransduction cascade:
Convert a photon of light into an electrochemical signal. RHO
Mutations in this gene are the most common single cause of RPRhodopsin
adRP, arRP
PDE6A and 6B
Phosphodiesertase alpha and beta subunits
arRP
SAG
Arrestin
arRP, arCSNB
CNGA1 and B1
Rod cGMP-gated channel alpha and beta subunits
arRP
GUCY2D and GUCA1A
Retina-specific guanylate cyclase and GC-activating protein
CRD, LCA and cone dystrophy
II. Visual cycle:
Metabolize retinol (vitamin A).RPE65
Retinal pigment epithelium-specific 65-kd protein
arRP, LCA
RLBP
Cellular retinaldehyde-binding protein
arRP, ar retinitis punctata albescens
ABCA4
ATP-binding cassette transporter
arRP, CRD, Stargardt disease
LRAT
Lecithin retinol acyltransferase
arRP
RGR
RPE-retinal G protein-coupled receptor
arRP
III. Structural proteins:
Maintain shape of the rod and outer-segment discs.RDS
Peripherin /RDS
adRP
RHO
Rhodopsin
adRP, arRP
IV. Transcription factors:
May control the actions of many photoreceptor-specific genes.CRX
Cone-rod otx-like homeobox transcription factor
adRP, LCA, arCRD
NRL
Neural retinal leucine zipper
adRP
NR2E3
Nuclear receptor subfamily 2 group E3
arRP
V. Cell-cell interactions:
May play a role in cell-to-cell receptor communication between the RPE and the photoreceptors.MERTK
C-mer proto-oncogene tyrosine kinase
arRP
CRB1
Crumbs homolog 1
arRP, LCA
USH2A
Usherin
arRP, Ushers syndrome
VI. Splicing factors:
Encode small proteins that help to produce messenger (m) RNA from pre-mRNA.PRPF8
Pre-mRNA
adRP
PRPF3
Processing factor
PRPF31
8, 3 and 31 respectively
VII. Intracellular transport:
Transport proteins from the photoreceptor inner segments to the outer segments.FSCN2
Retinal fascin
adRP
RP1
RP1 protein
adRP
RPGR
Retinitis pigmentosa
GTPase regulatorX-linked RP
RP2
RP2 protein
X-linked RP
TULP1
Tubby-like protein 1
arRP
Miscellaneous genes:
(no specific functional group)IMPDH1
Inosine monophosphate dehydrogenase 1
adRP
RP9
RP9 protein
adRP
adRP = autosomal dominant RP arRP = autosomal recessive RP
LCA = Leber congenital amaurosis CRD = cone/rod dystrophy
Leber Congenital Amaurosis
Leber congenital amaurosis (LCA) was first described 135 years ago by German ophthalmologist Theodore Leber as a group of in-herited disorders that caused severe vision loss from birth. Dr. Leber noted that although the fundus looked normal early on, it eventually acquired a degenerative, or mottled, appearance.
LCA affects about 5,000 children in the United States.1 These children present with nystagmus and severe visual impairment.
The diagnostic hallmark test re-sult of this disease is a markedly abnormal or flat ERG.5 This is an important test to order if you suspect LCA, as the fundus can look normal early on, or it can display a variety of unusual appearances.
So, regardless of family history, any child who presents with severely reduced visual acuity accompanied by nystagmus should have an ERG performed if possible. (Some-times, the child may not cooperate with the test, or you may have difficulty locating a facility that offers ERG testing.) The disease can be progressive and can lead to visual acuity of light perception.
LCA is usually inherited as an autosomal recessive trait. There are currently mutations on eight genes known to cause LCA. These are GUCY2D, RPE65, CRX, TULP1, AIPL1, CRB1, RP-GRIP and RDH12.6 In the United States, these account for only 10% of LCA cases. There is a complicated form of LCA that is accompanied by developmental delays, deafness and seizures. However, none of the above-listed genes is known to be associated with the complicated form.
One form of LCA is caused by mutations in the RPE65 gene. This protein is responsible for the conversion of vitamin A (all-trans-retinol) to 11-cis-retinol in the RPE, which is necessary to produce rhodopsin. Mutations in this protein therefore affect rhodopsin formation.
Research using RPE65-deficient mice, in which a substitute for rho-dopsin was administered, resulted in rapid recovery of the ERG and partial prevention of cone loss.7
Another therapy also been studied: gene replacement therapy, using adenovirus vectors. This was demonstrated to be successful in Briard dogs deficient in RPE65.8
The significance of having a reliable animal model to study LCA: This form of the disease will be the first retinal dystrophy for which there will be gene therapy clinical trials in humans.
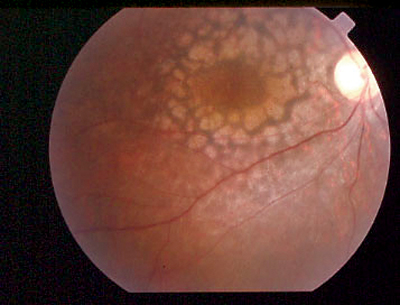 |
| Leber congenital amaurosis, with severely reduced visual acuity from birth and nystagmus, can look like anything. |
Stargardt Disease
Stargardt disease is a hereditary disease that affects the macula in young patients and was first described 97 years ago by Prof. Karl Stargardt. Prevalence is estimated to be between one in 8,000 and one in 10,000.9
In 1962, another central retinal dystrophy characterized by later-onset flecks was described by ophthalmologist Adolphe Franceschetti, which he termed fundus flavimaculatus. Eventually, clinicians recognized the association of these two separate findings as one disease, formerly referred to as Stargardt/fundus flavimaculatus. Inheritance is autosomal recessive, although there is a rare autosomal dominant Stargardt-like dystrophy.
The typical patient: a teenager who presents with a gradual loss of central vision. Early on, the fundus can look almost normal. Often there is a macular granularity that can eventually progress to cause a metallic, or beaten-bronze, appearance. Many patients also demonstrate deep yellowish fish-tail or pisciform flecks, made up of lipofuscin pigment. Some patients have maculopathy with no flecks, and some have flecks with no maculopathy. These can change in appearance over time and can spread to the mid-periphery as the patient ages. Color vision may be normal or mildly reduced as is the ERG, since this disease affects only the central retina.
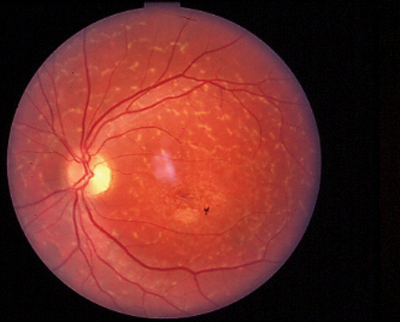 |
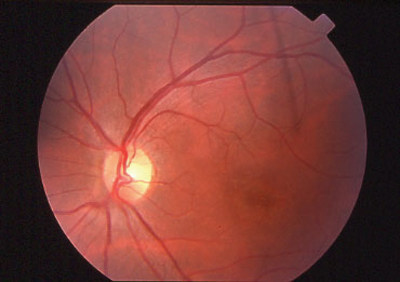 |
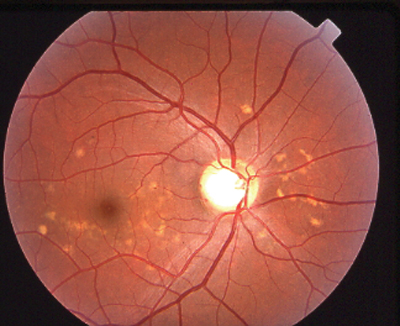 |
| Stargardt disease can present as a macular lesion with flecks (top), no flecks (middle), or flecks with no macular lesion (bottom). |
Fluorescein angiography helps the clinician diagnose Stargardt disease. It demonstrates areas of hyperfluoresecence in macular window defects (which are RPE disturbances) and a darkening of the choroidal flush, termed a dark or silent choroid.
Now, there is genetic proof that that Stargardt disease and fundus flavimaculatus are in fact the same disease, resulting from mutations in the same gene. This gene is ABCA4, a rather large gene on chromosome 1 discovered in 1997 by Rando Alikmets.10 The ABCA4 gene transports all-trans-retinal out of the photoreceptor outer segments during the visual cycle. Mutations in this gene affect this transport, allowing for the accumulation of all-trans-retinal, which eventually reacts with other substances to form certain products that get converted to a toxic vitamin-A based fluorophore, called A2E. Accumulation of A2E causes lipofuscin to build up, resulting in photoreceptor cell death.
Other ABCA4 Retinopathies
Mutations in the ABCA4 gene are also responsible for cone-rod dystrophy (a group of diseases that affect the cones and central vision first and then the rodsthe opposite of RP). This dystrophy causes reduced vision and color vision loss and eventually night vision problems. Clinically, the fundus can look normal or can have macular changes (see page 76). The pres-ence of multiple mutations in the ABCA4 gene are also responsible for some forms of RP. There are cases in which one member of the patients family has Stargardt disease and a close relative has RP.
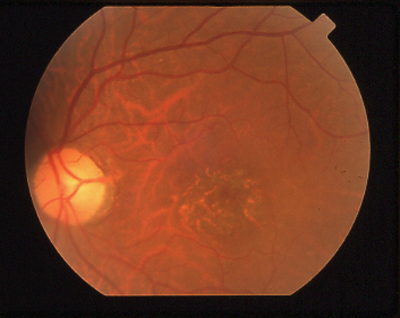 |
|
A granular macula with reduced visual acuity and abnormal color vision is characteristic of cone/rod dystrophy. |
ABCA4 mutations are also associated with 1% to 2% of age-related macular degeneration (AMD). Although ABCA4 mutations account for a small amount of AMD cases, A2E accumulation plays an important role in the pathogenesis of common forms of AMD.
Future treatment of these diseases, including AMD, may lie in the development of pharmacological agents designed to reduce the amount of A2E in the RPE. Re-search using 11-cis-retinoic acid demonstrated inhibition of RPE65 protein and subsequent decrease in A2E accumulation.11
Your Role
When a patient presents with a suspected hereditary disease, there are four things you should do:
Obtain a good case history. If you suspect RP and a patient does not complain of night vision problems, for instance, that patient may live in a city that has good lighting at night. One helpful question to ask is if the patient can find his seat in a movie theater as easily as others in his party.
Obtain a good pedigree. Find out who else in the family has been diagnosed with a retinal disease. Go back at least three generations, if possible, and be sure to inquire about cousins, related uncles and aunts, etc. Also, find out if there is parental consanguinity (are the parents related?), or if the patients parents grew up in the same small village with only a few thousand people. Both criteria tend to support autosomal recessive disease.
Order genetic testing if indicated. Contact medical genetic specialists in your area, and establish a co-managing relationship with them. (See Information About Genetic Tests and Clinical Trials, below.)
Be aware of ongoing genetic clinical trials. Several ongoing clinical trials are investigating new medical therapies for some of the retinal dystrophies. It is important to keep abreast of these should your patient be interested in and/or eligible for recruitment.
If you see relatively symmetric findings when examining the retina, think hereditary etiology.
Information About Genetic Tests and Clinical Trials Currently, research laboratories and commercial labs offer genetic testing for the hereditary eye diseases. For a relatively quick answer (eight to ten weeks), blood samples are sent to commercial laboratories to be tested for specific genetic mutations based on the suspected diagnosis. One such commercial laboratory: the Carver Laboratory for Molecular Diagnosis (www.carverlab.org) at the University of Iowa. The downside of commercial testing: Insurance may not cover the cost, and it can be quite pricey, meaning hundreds of dollars or more. Blood samples can also be sent to individuals who conduct genetic research of the retinal dystrophies (www.genetests.org). This service is free, supported by grant funding. However, results can take years. Keep in mind that we do not yet know all the genes responsible for many forms of the retinal dystrophies. Therefore, negative test results do not necessarily mean the patient will not develop the disease. In addition, positive results do not necessarily predict the phenotypic presentation. To find out more about ongoing clinical trials, go to www.ClinicalTrials.gov, enter the disease of interest, and find out what trials are recruiting patients. Your affected patients should be informed of these trials in case they are interested in participating.
Many retinal dystrophies are diagnosed based on symptoms, the clinical picture and testing results. Genetic testing is not yet the mainstay of clinical care, but it may be an option for patients who want to know if they or someone else in their family is or will be affected by a particular ocular disease. Some patients may want to know if their child will develop the disease or the likelihood that future progeny will be affected.
The retinal dystrophies can have devastating effects on vision and visual function. Many of these diseases can be managed with low-vision devices to some degree, but currently there is no approved medical treatment known to halt disease progression or restore vision. The future will likely change this.
Now that we know many of the genetic mutations of these diseases, we will surely witness over the next decade several forms of treatment, including gene replacement, retinal transplantation, cell rescue and the development of pharmaceutical agents. Many clinical trials are currently underway.
Dr. Bass is a distinguished teaching professor at the State University of New York State College of Optometry. She is an instructor in the posterior segment disease course in the professional program, and she teaches in the retinal clinic.
Next month: Dr. Bass discusses the genetics of glaucoma.
1. Weleber RG. Inherited and orphan retinal diseases: phenotypes, genotypes, and probable treatment groups. Clinical Progress in Inherited Orphan Retinal Diseases. Retina Supplement 2005 Dec;25(8): S4-S7.
2. Hims MM, Diager SP, Inglehearn CF. Retinitis pigmentosa: genes, proteins and prospects. In: Wissinger B, Kohl S, Langenbeck U (eds.): Genetics in Ophthalmology.
Developmental Ophthalmology. Vol. 37 Basel, Switzerland. Karger, 2003, 109-25.
3. Rivolta C, Sharon D, DeAngelis MM, Dryja TP. Retinitis pigmentosa and allied diseases: numerous diseases, genes and inheritance patterns. Hum Mol Genet 2002 May 15; 11(10):1219-27.
4. Maubaret C, Hamel C. Genetics of retinitis pigmentosa: metabolic classification and phenotype/genotype correlations. J Fr Ophthalmol 2005 Jan;28(1):71-92.
5. Francheschetti A, Dieterle P. Importance diagnostique at prognotique de lelectroretinogramme (ERG) dans les degenerescences tapeto-retiniennes avec retresissement du champ visual et hemeralopie. Confinia Neurologica 1954; 14:184-6
6. Allikmets R. Leber congenital amaurosis: a genetic paradigm. Ophthalmic Genet 2004 Jun;25(2):67-79.
7. Van Hooser JP, Aleman TS, He YG, et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci USA 2000 Jul 18;97:8623-8.
8. Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 2001 May;28:92-5.
9. Detman AF. Stargardt Disease. Orphanet Encyclopedia January 2003. www.orpha.net/data/patho/GB/ukStargardt . pdf. (Accessed April 5, 2006)
10. Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997 Sep;15:236-46.
11. Radu RA, Mata NL, Nusinowitz S, et al. Isotretinoin treatment inhibits lipofuscin accumulation in a mouse model of recessive Stargardts macular degeneration. Novartis Found Symp. 2004;255:51-63; discussion 63-7, 177-8.
