 |
|
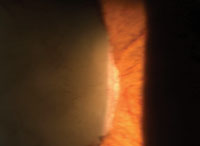
1. Sea-fan neovascularization associated with subhyaloid hemorrhage and retinal fibrosis.
|
 |
|
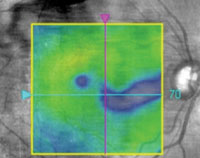
2. Paramacular thinning on OCT in the right eye.
|
 |
|
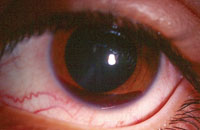
3. Iris neovascularization.
|
 |
|
4. Spontaneous hyphema.
|
 |
|
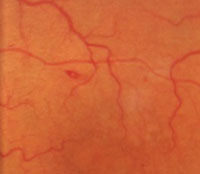
5. Sunburst.
|
 |
|
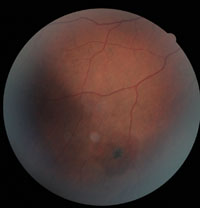
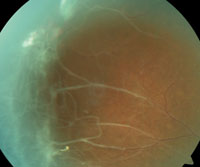
6. Roth spot hemorrhage.
|
 |
|
7. PSCR with neovascularization and fibrosis.
|
Sickle-cell disease (SCD), a hereditary autosomal recessive condition, is characterized by the presence of crescent- or sickle-shaped red blood cells. These anomalous cells prevent normal blood flow and promote increased blood viscosity, hypoxia, venous stasis, acidosis and inflammation associated with varied systemic and ocular complications.1
SCD is one of the most prevalent genetic disorders in the US, predominantly affecting people of African or Mediterranean descent.
More than two million people—about 8.5% of the US population of African ancestry—have sickle-cell trait, and as many as 100,000 African Americans currently have the condition.2
Here, we examine the cases of two black females who experienced ocular complications secondary to SCD.
Case #1
History
A 43-year-old black female presented with a chief complaint of foreign body sensation and burning that had persisted for three days. Her medical history was remarkable for sickle cell trait. Further, her ocular history was significant for a diagnosis of sickle-cell retinopathy in 2006.
Diagnostic Data
Her best-corrected visual acuity was 20/25-3 OD, 20/25-2 OS. Pupils were equal, round and reactive to light, with no evidence of relative afferent defect. Confrontation fields were full to finger counting in both eyes, and extraocular motility testing showed full range of motion.
Biomicroscopy evaluation revealed a yellowish scleral hue, as well as subconjunctival hemorrhages located near the inferior palpebral conjunctiva OU.
Intraocular pressure measured 17mm Hg OD and 18mm Hg OS. Dilated fundus examination revealed normal physiological optic nerve cupping with pink and distinct margins in both eyes. Both maculae were flat and intact, and her vessels exhibited a normal caliber OU.
Examination of the peripheral retina revealed the presence of intraretinal hemorrhages (salmon patches), arteriovenous anastomoses, sea-fan neovascularization and retinal fibrosis OD (figure 1), with temporal fibrosis located near an area of sea-fan neovascularization OS.
Diagnosis
We educated the patient on the findings, notified her primary care provider and referred her to a retina specialist for evaluation and possible treatment with anti-VEGF therapy. We scheduled a follow-up examination in four weeks.
Case #2
History
A 65-year-old black female presented with complaints of decreased vision in her right eye. Her medical history was remarkable for type 2 diabetes, hypertension and hypercholesterolemia––all of which were controlled with oral medications. Her family history was positive for sickle cell trait.
Diagnostic Data
Her best-corrected visual acuity was 20/30 OD and 20/25+ OS. Pupils and ocular motilities were unremarkable. Intraocular pressure measured 19mm Hg OU.
Dilated fundus examination revealed mild cataracts and non-proliferative diabetic retinopathy in both eyes. Optical coherence tomography (OCT) revealed no clinically significant macular edema, but we noted paramacular thinning in the right eye (figure 2). This warranted further evaluation; however, she was lost to follow-up.
Discussion
Sickle-cell disease is a complex disorder with distinct variants that occur when a single-point mutation of normal hemoglobin results in a sickle hemoglobin gene.3 Inheritance of this gene from both parents results in a homozygote condition known as sickle-cell anemia (HbSS). There are several heterozygous forms of SCD, including sickle-hemoglobin C disease (HbSC) and sickle beta-thalassemia (SThal).
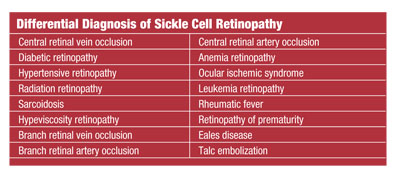
HbSC is associated with a substitution of lysine for glutamic acid, and SThal occurs from a point mutation in the beta globin subunit affecting the amino acid sequence. The most common variant is the carrier-state known as sickle-cell trait (HbAS) (see “Differential Diagnosis of Sickle Cell Retinopathy”).
Systemic and ocular manifestations vary. While sickle-cell anemia is associated with life-threatening systemic conditions, ocular complications are comparatively milder.4 On the other hand, SCD and SThal are associated with more severe ocular disease, yet have far fewer systemic manifestations. Further, the incidence of proliferative sickle-cell retinopathy in sickle-cell disease and thalassemia is higher than in sickle-cell anemia.1
It is important to note that a carrier of the sickle-cell trait may also experience both systemic and ocular complications.5
• Ocular findings. Resultant vaso-occlusive changes yield several anterior segment complications, such as the “conjunctival sickling sign,” which is characterized by capillary vessel segmentation. Iris changes manifest as atrophy with or without anterior or posterior synechiae. Additionally, the develpment of iris neovascularization (INV) could lead to secondary glaucoma and severe vision loss (figure 3).6,7
The presence of a spontaneous hyphema in black patients could be indicative of underlying SCD (figure 4). Even a small hyphema potentially could precipitate significant IOP increase and/or glaucomatous development.8 Hyphemic glaucoma is a direct result of blood cell obstruction within the trabecular meshwork, as well as pupillary block secondary to large blood clots.9 A sickle-cell patient who presents with hyphema is at risk for rebleed, and should be educated and monitored closely.
Although glaucoma medications may be required when treating a patient with hyphema and concomitant increase in IOP, the use of acetazolamide is contraindicated in SCD patients. This is related to the CAI effects on lowering pH as well as increasing ascorbate concentration in the aqueous, promoting sickling
of red blood cells in the anterior chamber, which may obstruct the trabecular meshwork.
Posterior segment findings include optic neuropathies, retinopathies, maculopathies, retinal hemorrhages, choroidopathy, vascular changes associated with tortuosity, “silver-wire” arterioles, angioid streaks, and arterial and vein occlusions.10
Given the spectrum of posterior ocular findings, you must consider several differential diagnoses.
The clinical features of sickle-cell retinopathy may be either nonproliferative or proliferative. Nonproliferative retinal changes include intraretinal salmon-patch hemorrhages, which may develop into a “black sunburst” upon resolution (figure 5). Iridescent spots, representing a collection of hemosiderin-laden macrophages, also may be noted.
 |
 |
Other retinal manifestations and vasculopathies also may be noted in patients with SCD. Although a Roth’s spot hemorrhage may be associated with bacterial endocarditis, it may also occur in patients with SCD (figure 6). Its presence should alert the physician for possible underlying vascular and hyperviscosity conditions, including SCD.
Angiopathies are also common, and venous tortuosity may be observed in up to 47% of HbSS and 32% of HbSC patients.11 Angioid streaks, a defect within Bruch’s membrane, can be seen in 1% to 2% of patients with SCD and warrant an OCT evaluation to rule out associated choroidal neovascular membrane (CNVM).12-14
In addition to the aforementioned findings, the appearance of a dark hyphema in the absence of increased intraocular pressure has been linked to a diagnosis of SCD since the mid-1970s.15
Proliferative sickle-cell retinopathy is initially characterized by the presence of arterial occlusion, leading to arteriovenous anastomoses (see “Proliferative Retinopathy: Goldberg Classification”). This is in response to localized peripheral ischemia from repeated episodes of arteriolar closure, which is presumed to trigger angiogenesis through the production of endogenous vascular growth factors.22 Disease progression leads to the formation of sea-fan neovascularization (as was documented in Case #1), fibrosis (figure 7), vitreous hemorrhage and tractional retinal detachment.
• New developments. Given the direct effect of SCD on the retinal vascular pathway, an array of maculopathies—including hairpin-shaped venular loops and irregularities of the foveal avascular zone—have been reported.21 The advent of OCT has led to better evaluation of macular morphological changes associated with SCD. Choroidal neovascular membrane occurs in 72% to 86% of cases with angioid streaks. If left untreated, the visual prognosis for SCD patients who present with angioid streaks is poor.27-29 Affected individuals should undergo an OCT evaluation.
Ancillary OCT has demonstrated a recent finding associated with macular (paramacula) thinning.16,17 A study using spectral-domain optical coherence tomography (SD-OCT) showed correlating macular thinning in about 50% of eyes with SCD.18 This may be a direct result of chronic ischemia affecting the retinal ganglion cells and retinal nerve fibers as they course temporally toward the optic nerve (as seen in Case #2).19
Macular ischemia typically develops along the temporal horizontal raphe, sparing the fovea and central visual acuity.20
• Lab analysis. It is helpful to know which laboratory tests most frequently are used to diagnose sickle-cell anemia and trait. Consider ordering lab tests if you observe the following:
- Dark hyphema without increased intraocular pressure or angioid streaks in black or Caribbean patients.
- Retinal vasculopathies in the absence of other associated systemic-related diseases.
- Presence of salmon-patch hemorrhages, sunbursts or comma-shaped conjuctival vessels.
A complete blood count (CBC) is used to evaluate a wide range of hyperviscosity-related disorders, including anemia, infection and leukemia.
A CBC with differential measures several components and features of blood, including red and white blood cells, hemoglobin, hematocrit and platelets––all of which may be abnormal in patients with SCD.
Hemoglobin electrophoresis is used to measure differences in the distinct oxygen-carrying protein. There are a number of different types of hemoglobin associated with SCD. A sickle cell test looks for abnormal hemoglobin in the blood, causing sickle-cell anemia. This test is run with concomitant abnormal hemoglobin.
• Management. Conservative management with close observation is warranted for nonproliferative SCR; however, various treatment modalities are frequently employed for proliferative SCR.
Patients who present with stage 3 to 5 proliferative disease should be referred to a retina specialist due to the potential risk of severe vision loss.
Conventional treatment includes panretinal photocoagulation.
Additionally, anti-VEGF therapy is regarded as a potential treatment option for proliferative disease.24-26
Our role as eye care providers is multidisciplinary. A patient may present with the established diagnosis of sickle-cell disease, which will require continuous communication with the PCP. The patient’s PCP should be made aware of all findings––even in the absence of any ocular manifestations. Moreover, we should be diligent in obtaining accurate case histories in SCD patients.
We must be familiar with the lab analysis used to diagnose sickle-cell anemia in order rule out other disease states and more effectively comanage the patient with his or her PCP.
Further, we must be able to recognize the presence of proliferative SCR and make a swift referral to prevent severe vision loss.
Dr. Demeritt is an assistant professor and attending optometrist at Nova Southeastern College of Optometry/The Eye Care Institute.
Dr. Shechtman is an associate professor of optometry at Nova Southeastern University College of Optometry, where she serves as an attending optometric physician at the Eye Institute and Diabetic/Macula Clinic.
Dr. Reynolds is an Associate Professor at Nova Southeastern University College of Optometry, where she is a clinical attending in the diabetes clinic and is a member of the Optometric Retina Society.
1. Emerson GG, Harlan JB, Fekrat S, et al. Hemoglobinopathies. In: Ryan SJ, ed. Retina. 4th ed. Philadelphia: Elsevier; 2006.
2. Sickle Cell Disease: Data & Statistics. Centers for Disease Control and Prevention. Available at: www.cdc.gov/ncbddd/sicklecell/data.html. Accessed July 2013.
3. Schnog JB, Duits AJ, Muskiet FA, et al. Sickle cell disease; a general overview. Neth J Med. 2004 Nov;62(10):364-74.
4. Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease: Cooperative Study of Sickle Cell Diseae. Blood. 1995 Jul 15;86(2):776-83.
5. Sowka J, Gurwood A, Kabat A. Sickle Cell Disease. Handbook of Ocular Disease Management. Rev Optom. Suppl.
6. Serjeant GR. The sickle cell trait. In: Serjeant GR (ed.). Sickle Cell Disease, 2nd Ed. New York: Oxford University Press; 1992.
7. Clarkson JG. The ocular manifestations of sickle-cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc. 1992;90:481-504.
8. Goldberg MF. Sickled Erythrocytes, hyphema, and secondary glaucoma: I. The diagnosis and treatment of sickled erythrocytes in human hyphemas. Ophthalmic Surg. 1979 Apr;10(4):17-31.
9. Gottsch JD. Hyphema: diagnosis and management. Retina. 1990;10 Suppl 1:S65-71.
10. Sayag D, Binaghi M, Souied EH, et al. Retinal photocoagulation for proliferative sickle cell retinopathy: a prospective clinical trial with new sea fan classification. Eur J Ophthalmol. 2008 Mar-Apr;18(2):248-54.
11. Welch RB, Goldberg MF. Sickle cell hemoglobin and its relation to fundus abnormality. Arch Ophthalmol. 1966 Mar;75(3):353-62.
12. Clarkson JG. The ocular manifestations of sickle cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc. 1992;90:481-504.
13. Condon PI, Serjeant GR. Ocular findings in elderly cases of homozygous sickle cell disease in Jamaica. Br J Ophthalmol. 1976 May;60(5):361-4.
14. Nagpal KC, Asdourian G, Goldbaum M, et al. Angioid streaks and sickle hemoglobinopathies. Br J Ophthalmol. 1976 Jan;60(1):31-4.
15. Nagal KC, Goldberg MF, Asdourian G, et al. Dark-without-pressure fundus lesions. Br J Ophthalmol. 1975 Sep;59(9):476-9.
16. Nagpal KC, Goldberg MF, Rabb MF. Ocular Manifestations of sickle hemoglobinopathies. Surv Ophthalmol. 1977 Mar-Apr;21(5):391-411.
17. Acacio I, Goldberg MF. Peripapillary and macular vessel occlusion in sickle cell anemia. Am J Ophthalmol. 1973 May;75(5):861-6.
18. Chou FY, et al. Hemoglobinopathies [ARVO abstract 3554]. Invest Ophthalmol Vis Sci. 2010;51.
19. Murthy RK, Grover S, Chalam KV. Temporal Macular Thinning on Spectral-Domain Optical Coherence Tomography in Proliferative Sickle Cell Retinopathy. Arch Ophthalmol. 2011 Feb;129(2):247-9.
20. Jampol LM. Arteriolar occlusive diseases of the macula. Ophthalmology. 1983 May;90(5):534-9.
21. Joussen AM, Gardner TW, Kirchhof B, Ryan AJ (eds.). Retinal Vascular Disease. Berlin: Springer; 2007.
22. Gitter KA. Dominantly inherited peripheral retinal neovascularization. Arch Ophthalmol. 1978 Sep;96(9):1601-5.
23. Jampol LM, Goldbaum MH. Peripheral proliferative retinopathies. Surv Ophthalmol. 1980 Jul-Aug;25(1):1-14.
24. Shaikh S. Intravitreal bevacizumab (Avastin) for the treatment of proliferative sickle retinopathy. Indian J Ophthalmol. 2008 May-Jun;56(3):259.
25. Siqueira RC, Costa RA, Scott IU, et al. Intravitreal bevacizumab (Avastin) injection associated with regression of retinal neovascularization caused by sickle cell retinopathy. Acta Ophthalmol Scand 2006;84:834-5.
26. Siqueira RC, Costa RA, Scott IU, et al. Intravitreal Bevacizumab (Avastin) Injection associated with regression of retinal neovascularization caused by sickle cell retinopathy. Acta Ophthalmol Scand. 2006 Dec;84(6):834-5.
27. Piro PA, Scheraga D, Fine SL. Angioid Streaks natural history and visual progression. In: Fine SL, Owens SL (eds.). Management of Retina Vascular and Macular Disorders. Baltimore: Williams and Wilkins; 1983.
28. Mansour AM, Shields JA, Annesley WH, et al. Macular degeneration in angioid streaks. Ophthalmologica. 1988;197(1):36-41.
29. Singerman LJ, Hatem G. Laser treatment of choroidal neovascular membranes in angioid streaks. Retina. 1981;1(2):75-83.
