 |
We all know the classic presentation of uveitis: cells and flare, pain, photophobia and reduced vision. But because a multitude of events can trigger such a response, we sometimes lose sight of how these processes occur and what they signify about our patient. Myriad underlying systemic conditions can cause anterior uveitis (AU); it is often the ocular sequela of an autoimmune condition or infection, and many of the potential causes lead to significant morbidity if left unmanaged.1-3 The patient’s systemic and ocular health rely on you, the optometrist, to carefully and strategically undertake a systemic review and work-up of AU patients, to investigate and potentially uncover, an infectious, autoimmune or inflammatory underlying etiology.
Historically, the majority of AU cases were thought to be idiopathic; now we know that a properly executed work-up, combined with more recent and advanced diagnostic techniques, yields an underlying systemic diagnosis in 70% of cases.4 The need is clear for a careful ocular evaluation and review of histopathological and immunological mechanisms to further narrow down the systemic work-up. This month, we will focus on the steps to achieve this.
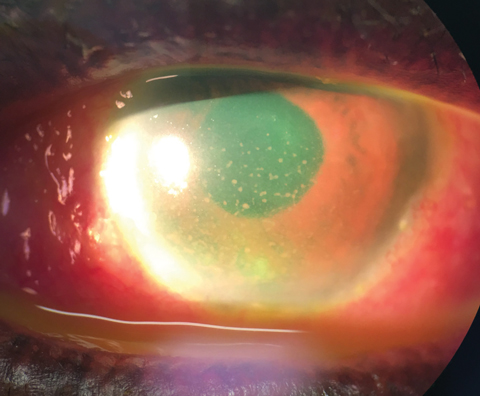 |
| Deposition of keratic precipiates on the inferior corneal endothelium. |
AU Common Symptoms
The most commonly reported symptom of AU is blurred vision, caused by the infiltration of cells into the otherwise clear anterior chamber and anterior vitreous cavity.1,2,5,6 In cases of Fuchs’ uveitis syndrome (FUS), blur is often the only reported symptom; this is a key feature to help you differentiate between FUS and other types of AU. For FUS, treatment with corticosteroids is sufficient and work-up is unnecesary.5,6
Pain is a common complaint of AU with an as-yet undisclosed systemic etiology. Pain as a response to light, and described as worsening at near, is caused by ciliary and iris sphincter muscle spasms.1,2 It can vary in severity and may be described as midly as a dull ache or as extreme as referred pain that spans the area supplied by the trigeminal nerve.2
Tearing, a result of trigeminal nerve stimulation and irritation, is another commonly reported symptom.2 Other possible symptoms include redness and, less commonly, floaters.2
Exam Intricacies
AU presents, anatomically, in three forms:
• iritis (involving only the iris);
• anterior cyclitis (anterior portion of the ciliary body); and
• iridocyclitis (a combination of iris and anterior ciliary body).1-3
Though the clinical findings that lead to a general diagnosis of AU are fairly similar, a few key intricacies exist that can help determine the subsequent systemic questionnaire and work-up, after you’ve confirmed a case.
Table 1. Systemic Etiologies of Uveitis | |
| Granulomatous | Non-ganulomatous |
| Herpes viruses: simplex, zoster, cytomegalovirus, Epstein-Barr Sarcoidosis Tuberculosis Lyme disease Syphilis Multiple sclerosis | HLA-B27 conditions: ankylosing spondylitis, Reiter’s syndrome, Crohn’s disease, ulcerative colitis Behcet’s syndrome Juvenile idiopathic arthritis Tubulointerstitial nephritis |
Conjunctival injection. The dilation of the episcleral blood vessels in response to inflammation causes prominent vasculature and hyperemia to appear around the limbus.1,4 The conjunctival injection may also be diffuse.1,2 Both FUS and Posner-Schlossman syndrome (PSS) present with low-grade inflammation, and these diagnoses should be considered in the absence of prominent conjunctival injection.5-7
Keratic precipitates (KPs). These deposits of various cell types on the surface of the corneal endothelium serve as perhaps the most important clinical finding in narrowing the potential cause of a patient’s AU. Fine, stellate and diffuse KPs are made up of lympho-plasmocytic inflammatory cells and indicate a non-granulomatous etiology, which may be allergic inflammatory in nature; on the other hand, medium to large “mutton fat” KPs consist of both lympho-plasmocytic inflammatory and epitheloid cells and point to granulomatous inflammation.1,2,4,8 Pigmented KPs are often indicative of a previous episode or episodes of AU, in contrast to fresh KPs that appear round, white and fluffy.1-3
Paying close attention to the exact location and deposition of KPs helps distinguish some conditions. The normal convection current of the aqueous is created by a temperature gradient, where cells are moved upwards towards the lens where the temperature is warmer, and then downwards towards the cornea where the temperature is cooler, depositing inferiorly on the endothelium.2 The pattern of this deposition, known as “Arlt’s triangle,” is displayed as a base-down triangle at the inferior corneal endothelium.2,3 KPs deposited outside of this triangle, in a diffuse pattern, are suggestive of FUS and herpetic etiologies.2,3,5,9
Anterior chamber cells and flare. This clinical manifestation is pathognomonic for AU. Aqueous cells consist of inflammatory cells (mainly lymphocytes, and neutrophils to a lesser degree) that have leaked into the anterior chamber as a result of the breakdown in the blood-aqueous barrier.1,2,4 These cells are amelanotic and must be differentiated from the brownish pigment that is released in cases of pigment dispersion syndrome or following pupillary dilation.4 The exudation of protein (albumin) into the anterior chamber comprises flare.2
The quantity of cells and flare in the anterior chamber is helpful in differentiating purely ocular vs. systemic etiologies. Systemic causes of cells or flare are typically more significant, as they are caused by more severe extraocular inflammatory diseases. If seen in trace quantities, PSS and FUS should be excluded before assuming systemic work-up.5-7 By contrast, if cells are seen in dense quantities, forming a hypopyon or fibrinous clot, these typically suggest AU secondary to HLA-B27-related conditions such as juvenile idiopathic arthritis (JIA) or Behcet’s disease.1,4
Iris changes. Inflammatory cells may also deposit onto the surface of the iris in granulomatous forms of uveitis. When they are located on the pupillary border, they are called Koeppe’s nodules. These nodules, when located elsewhere on the iris body stroma, are called Busacca’s nodules.1,4
Iris atrophy may also be seen in FUS and AU that is secondary to herpetic disease. Diffuse iris atrophy and hypochromia are seen more commonly in FUS, whereas sectoral atrophy is more characteristic of AU caused by the herpes virus.5,6,9 Though the exact mechanism is unknown, researchers believe that the herpes virus invades the pigment epithelium of the iris, causing these atrophic patches in previous or active bouts of AU.10 In contrast, researchers believe the etiology of FUS (also not completely understood) is a sympathetic dysfunction; as such, the hypochromic iris—indicating the affected eye—may be a result of decreased innervation to the iris stroma.11
Intraocular pressure (IOP). In AU, patients may present with abnormal IOP, which can be either significantly higher or lower than the non-affected eye. Unilateral IOP spikes on initial presentation are characteristic of herpes simplex/zoster or PSS.4,7,9 Studies show both the herpes virus and PSS to cause damage or inflammation that is localized to the trabecular meshwork, which explains the elevation in IOP.4,7,9 This is in contrast to lower IOP in the affected eye, which is more suggestive of an etiology, such as HLA-B27-related uveitides from ciliary body ischemia and less aqueous production.4
Table 2. Summary of Clinical Features of PSS and FUS | ||
| Posner-Schlossman | Fuchs’ Uveitis Syndrome | |
| Symptoms | Blur or haloes No pain | Blur or haloes No pain |
| Signs | Unilateral presentation Low grade cells Fine KPs No injection Significantly elevated IOP | Unilateral presentation Low grade cells Diffuse fine or mutton fat KPs Iris hypochromia/diffuse atrophy No injection |
| Treatment | Anti-hypertensives Corticosteroids No systemic work-up indicated | Not usually responsive to corticosteroids No systemic work-up indicated |
Putting the Pieces Together
Carefully examining the clinical manifestations on initial presentation of AU alone can significantly reduce the clinician’s list of differential diagnoses. This also eliminates the “shotgun” approach of non-systematic, overly laborious medical work-up; rather, it promotes a more targeted approach to help determine a management plan. Of course, given the possible, and often probable, systemic underpinnings, anticipate that this plan will be carried out in conjunction with the patient’s primary care physician or rheumatologist.
| 1. Agrawal RV, Murthy S, Sangwan V, et al. Current approach in diagnosis and management of anterior uveitis. Ind J Ophthalmol. 2010;58(1):11-19. 2. Kelkar AS, Arora ER, Sowkath B, et al. Uveitis: classification, etiologies, and clinical signs. Del J Ophthalmol. 2016;26(4):264-71. 3. Guney E, Tugal-Tutkun I. Symptoms and signs of anterior uveitis. Touch Medical Media. 2013;6(1):33-7. 4. Herbort CP. Appraisal, work-up and diagnosis of anterior uveitis: A practical approach. Middle East Afr J Ophthalmol. 2009;16(4):159-67. 5. Nalcacioglu P, Ozdel PC, Simsek M. Clinical characteristics of fuchs’ uveitis syndrome. Turk J Ophthalmol. 2016;46:52-7. 6. Mocan MC, Kadayificilar S, Irkec M. In vivo confocal microscopic evaluation of keratic precipitates and endothelial morphology in Fuchs’ uveitis syndrome. Eye. 2012;26(1):119-25. 7. Green RJ. Posner-Schlossman syndrome. Clin Exp Optom. 2007;90(1):53-6. 8. Kanavi MR, Soheilian M. Confocal scan features of keratic precipitates in granulomatous versus nongranulomatous uveitis. J Ophthalmic Vis Res. 2011;6(4)255. 9. Kardes E, Bozjurt K, Akcay BI, et al. Clinical features and prognosis of herpetic anterior uveitis. Turk J Ophthalmol. 2016;46(3):109-13. 10. Marsh RJ, Easty DL, Jones BR. Iritis and iris atrophy in herpes zoster ophthalmicus. Am J Ophthalmol. 1974;78(2):255-61. 11. Mohamed Q, Zamir E. Update of Fuchs’ uveitis syndrome. Curr Opin Ophthalmol 2005;16(6):356-63. |
