 |
History
A 59-year-old male presented to the office with a chief complaint of blurry vision, with and without his correction at distance and near, in his right eye for three weeks. He denied having any previous ocular injuries or surgeries. His systemic history, however, was remarkable for a 10-year history of hypertension, appropriately managed with lisinopril. The patient reported no history of allergies of any kind.
Diagnostic Data
His best-corrected entering visual acuities were 20/50 OD and 20/20 OS at distance and near with no improvement upon pinhole. His external examination was normal with smooth extraocular muscle movements, normal color vision, full confrontation visual fields and no evidence of afferent pupil defect. Biomicroscopic examination of the anterior chamber found normal structures and tissues with open angles and normal intraocular pressures measuring 15mm Hg OU via Goldmann applanation.
The pertinent posterior segment finding—pictured—was discovered during dilated fundus exam.
Your Diagnosis
Does this case require additional tests? How would you manage this patient? What is the likely prognosis?
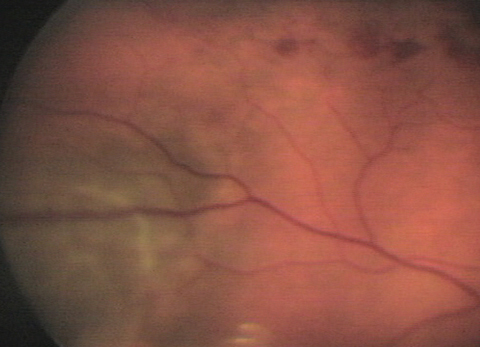 |
| This 59-year-old hypertensive patient’s right eye shows findings that may explain his blurry vision. Can you diagnose him? Click image to enlarge. |
Diagnosis
Additional testing included a peripheral visual 30 degree to 60 degree screening field, photography, B-scan ultrasonography and optical coherence tomography (OCT). A referral to retinology was made to rule out fluorescein angiography or additional neuroimaging.
The diagnosis is this issue is serendipitous discovery of choroidal melanoma (CM) during a dilated fundus examination following the patient’s presentation of symptoms produced by a resolving non-ischemic branch retinal vein occlusion in the right eye.
Choroidal melanoma is primary malignancy of the eye. It most commonly affects the choroid, but also can affect the iris (associated with a better prognosis), ciliary body (associated with a worse prognosis) or conjunctiva.1-3 Choroidal melanoma has a mortality rate of up to 50% due to hematogenous metastasis to the liver and a current lack of effective systemic therapy.1-5 Although rare (5 to 6 per 1 million), choroidal melanoma is the most common ocular cancer in adults.1,2,6 It has a predilection for fair-skinned, lightly pigmented races of European descent.1,2,4-7 Asian and Hispanic races have intermediate risk while African races show a low risk.6 Given the pathological means to cellular alterations the role of pigment in risk-determination suggests exposure to ultraviolet light as a causative factor.1,4,7 However, this connection has yet be proven.1,4,7 Published risk factors include oculodermal melanocytosis (nevus of Ota), the presence of cutaneous, iris, or choroidal nevi and age (mean age of diagnosis is 55 years).1,2,6
Choroidal melanoma is a neoplasm of uveal melanocytes that acquire some variation of pro-malignant cytogenetic mutations. These cytogenetic mutations give rise to specific phenotypic, histopathologic characteristics, which enable the identification of prognostic factors which influence the management plan.1 By far, the most significant cytogenetic prognostic factor is the loss of one copy of chromosome 3. Found in 50% of uveal melanomas, this finding—among others—is associated with increased risk of metastasis (70% with versus 20% without).1,2,4,5,6,8 The end result, in most cases, is increased malignancy and genetic instability. Researchers theorize that the altered gene expression and modified immune response feed into each other, creating a pro-tumor microenvironment that leads to local growth and systemic metastasis.3 Further understanding the role of gene expression and inflammation in choroidal melanoma is driven by the hope of using the pathway as an alternative route for treatment.3
Despite the recent advances in understanding of the genetic and inflammatory characteristics of choroidal melanoma, diagnosis is primarily based on the ophthalmoscopic appearance of the tumor. Therefore, efforts in enhancing the diagnostic accuracy of choroidal melanoma has predominantly focused on assessment via ophthalmoscopic appearance.1,5,6,7
Choroidal melanoma is usually seen as a dark, dome-shaped lesion but can take on a mushroom shape if it ruptures Bruch's membrane. Theses tumors have the ability to extend further into the globe.1,4 Shields et al.11 have produced a helpful mnemonic: “To Find Small Ocular Melanomas, Use Helpful Hints Daily” to group the ophthalmoscopic characteristics which distinguish a choroidal melanoma from a benign nevus.6,11 These characteristics are:
- Thickness >2mm,
- subretinal Fluid,
- Symptoms (present in 30% and include blurred vision, visual field defect, photopsia, irritation and pain),1
- Orange pigmentation,
- lesion Margin within 3mm of the optic disc,
- Ultrasonographic Hollowness,
- Halo of depigmentation around the lesion
- Drusen absent.6,11
These features can be observed with indirect biomicroscopy and B-scan ultrasonography. Flourescein angiography (FA) and OCT can also be used to confirm the diagnosis. OCT and automated perimetry are often used to assess the presence of an exudative retinal detachment which is a common cause of symptoms in choroidal melanoma.1,6 Ultrasonography can also be used to assess choroidal excavation or shadowing within the orbit which suggests the presence of orbital extension. Fine-needle aspiration cytology can be done but is controversial because of the theoretical potential for the seeding of tumor cells. Since the procedure is not necessary for accurate diagnosis it remains a controversial step.6
Growth over time is the final and most telling diagnostic and prognostic indicator. The observation of rapid growth over time clearly differentiates a choroidal melanoma from a benign nevus which will only grow slowly at a steady rate of 0.5mm per year.11 In a multivariate analysis of small suspicious choroidal melanocytic tumors, those with none of the above risk factors grew in only 4% of patients.12 Those with one risk factor experienced growth in 36% of patients, two risk factors showed growth in 45%, three and four risk factors showed growth in 50% and 51% respectively and five risk factors showed growth in 56% of tumors.12 The most significant combination of risk factors was a thickness >2mm, the margin of lesion being within 3mm of the optic disc and the discovery of the presence of subretinal fluid overlying the tumor. These factors are associated with a 63% chance of rapid growth.12
Conditions that can masquerade as a choroidal melanoma include choroidal nevus, other choroidal metastasis, choroidal hemangioma and retinal pigment epithelium (RPE) hypertrophy or hyperplasia. The presence of hemorrhage should also raise suspicion for an alternative diagnosis as this is rare in choroidal melanoma. CM are rarely defined by dark black pigmentation. In most instances lesions with this coloration are RPE hypertrophy or hyperplasia. An amelanotic lesion is characteristic of a metastasis or hemangioma.6
Once a diagnosis of choroidal melanoma has been made, the likelihood of metastasis must be assessed so that an appropriate local or systemic approach can be chosen. Tumor basal diameter (TBD) is the most important prognostic factor and is the primary tumor characteristic used to guide treatment.2,5,7 Tumor location also plays a role as ciliary body tumors have an increased risk for metastasis.2 Cytogenetic and histopathologic evaluation are also helpful.2,5,6 With monosomy 3, lymphocytic infiltration, homogenous epithelioid cell appearance, vascular loops and increased mitotic activity all associated with increased risk of metastasis.2,4
Treatment ranges from exenteration (removal of globe and all orbital contents) to radiation (external beam or insertion of a radioactive plaque [brachytherapy]).1 The Collaborative Ocular Melanoma Study (COMS) has provided guidelines for treatment of tumors based on size categorization.7 For suspicious nevi or small melanomas (1-3mm thick with >5mm diameter) the standard of care is observation; every 6 months for growth at first and then every year if no risk factors are present. If there are 1-2 risk factors, monitoring should be scheduled every 4-6 months. If 3 or more risk factors are present, the patient should be referred to an ocular oncology specialist to rule out treatment. If a decision is made to treat a small tumor, options include brachytherapy, laser photocoagulation, photodynamic therapy or external beam charged particle radiation.7,11 Tumor resection and enucleation are always included within the spectrum of treatment due to the ever-present danger of metastasis.7,11 Medium size tumors (3.1mm to 8mm thick and <16mm largest diameter) should be treated with either brachytherapy or enucleation.7,11 The COMS showed that brachytherapy, an eye-sparing treatment, showed no difference in mortality compared with enucleation.7,11 Therefore, treatment has shifted in favor of brachytherapy. Large tumors (>8mm thick and >16mm largest diameter) are predominantly treated with enucleation.7 Exenteration is considered when extension of the tumor moves into the orbit.7 There has been some suspicion that enucleation can cause increased incidence of metastasis as the tumor is disturbed through removal. To prevent this, radiation treatment prior to enucleation has been employed. The COMS, showed that there is no difference in mortality between enucleation and radiation plus enucleation.4,7
Overall, brachytherapy is the most common treatment for choroidal melanoma in the US.1,13 The radioactive plaque is inserted surgically and allowed to irradiate the tumor for 3-7 days before it is taken out. The plaque is usually shaped to be larger than the tumor's largest diameter by 2mm to ensure proper irradiation of the entire mass.6 The plaque material that is usually used is Iodine-125 because of its good tissue penetration, low energy and commercial availability. Gold-198, Strontium-90, Ruthenium-106 and Palladium-103 can also be used.6,14 Complications of brachytherapy can be sight threatening. They include radiation retinopathy, cataract, vitreous hemorrhage and neovascular glaucoma.6 These side effects should be discussed before proceeding. Other factors include cosmetic appearance and patient anxiety about tumor recurrence.7,15,16
Even with correct treatment, prognosis of choroidal melanoma is grim. Five year survival rates are 69% to 81.6% and ten year survival rates are 57% to 62%.1,3,5 After detection of metastases, long term survival is rare with mortality rates at 80% within one year and 92% in two years.1-3,5 A small cohort in the COMS with medium sized tumors forewent treatment. They were observed and showed a five year mortality rate of 30%. This was higher than the enucleation (19%) or brachytherapy (18%) cohorts.7 This clearly demonstrates the benefit of therapy.2,7
There has been a marked improvement in eye-sparing techniques in the treatment of choroidal melanoma.2 Unfortunately, the efforts have failed to create an improvement in survival rates.1-3,6 Less than 4% of patients with uveal melanomas have detectable metastases when they are diagnosed, however, eventually half will develop metastases.1,2,4 Most metastases begin before primary treatment occurs, underscoring the need to develop early recognition.4,17
Dr Gurwood thanks Marc Myers and Nicholas Karbach, OD for contributing this case.
| 1. Jovanovic P, Mihajlovic M, Djordjevic-Jocic J, et al. Ocular melanoma: an overview of the current status. International Journal of Clinical and Experimental Pathology. 2013;6(7):1230-1244. 2. Kaliki S, Shields CL, Shields JA. Uveal melanoma: Estimating prognosis. Indian Journal of Ophthalmology. 2015;63(2):93-102. 3. Bronkhorst IHG, Jager MJ, Uveal Melanoma: The Inflammatory Microenvironment. J Innate Immun 2012;4:454-462 4. Eagle RC. The pathology of ocular cancer. Eye. 2013;27(2):128-136. 5. Pereira PR, Odashiro AN, Lim L-A, et al. Current and emerging treatment options for uveal melanoma. Clinical Ophthalmology (Auckland, NZ). 2013;7:1669-1682. 6. Singh P, Singh A. Choroidal melanoma. Oman Journal of Ophthalmology. 2012;5(1):3-9. 7. Margo CE. The Collaborative Ocular Melanoma Study: An Overview. Cancer Control. 2004;11(5):304-9. 8. McCannel TA. Fine-needle aspiration biopsy in the management of choroidal melanoma. Curr Opin Ophthalmol. 2013;24(3):262-6. 9. Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;15;64(20):7205-9 10. Kusmartsev S, Gabrilovich DI. Role Of Immature Myeloid Cells in Mechanisms of Immune Evasion In Cancer. Cancer immunology, immunotherapy : CII. 2006;55(3):237-245. 11. Shields CL, Furuta M, Berman EL, et al. Choroidal Nevus Transformation Into Melanoma: Analysis of 2514 Consecutive Cases. Arch Ophthalmol. 2009;127(8):981-987. 12. Shields CL, Cater J, Shields JA, et al. Combination of clinical factors predictive of growth of small choroidal melanocytic tumors. Arch Ophthalmol. 2000;118(3):360-4 13. Robertson DM. Changing concepts in the management of choroidal melanoma. Am J Ophthalmol. 2003;136(1):161-70 14. Stannard C, Sauerwein W, Maree G, Lecuona K. Radiotherapy for ocular tumours. Eye. 2013;27(2):119-127. 15. Melia M, Moy CS, Reynolds SM.Quality of life after iodine 125 brachytherapy vs enucleation for choroidal melanoma: 5-year results from the Collaborative Ocular Melanoma Study: COMS QOLS Report No. 3. Arch Ophthalmol. 2006;124(2):226-38. 16. Damato B, Heimann H. Personalized treatment of uveal melanoma. Eye. 2013;27(2):172-179. 17. Eskelin S1, Pyrhönen S, Summanen P, et al.Tumor doubling times in metastatic malignant melanoma of the uvea: tumor progression before and after treatment. Ophthalmology. 2000;107(8):1443-9. |
