Multifocal pattern dystrophy (MPD) is one of the five types of autosomal dominant pattern dystrophies. These dystrophies are rooted in an inherited mutation on the peripherin/retinal degeneration slow (RDS) gene.1,2 The onset of the presentation of autosomal dominant pattern dystrophies is typically midlife; multifocal pattern dystrophy more specifically presents between the fourth and sixth decades.3-7 Autosomal dominant pattern dystrophies are characterized by normal to mild visual impairment and some patients report mild visual disturbances. Visual disturbances and deficits typically progress very little, with vision remaining mostly intact throughout life. While mild visual impact is typical, some cases report late vision loss secondary to atrophy, choroidal neovascularization or both.1,8-10 MPD typically presents with bilateral and symmetrical findings and shows as scattered, yellow, triradiate, fleck-like lesions in the posterior pole and arcades in the fundus.3,5,11 The following report will discuss a case of multifocal pattern dystrophy, the differentials to consider, additional workup and management.
 | |
| Fig. 1. These fundus images of a 58-year-old black male with multifocal pattern dystrophy in both eyes show a triradiate configuration of flecks within the posterior pole, extending just beyond the vascular arcades. |
Case Report
A 58-year-old black male was referred to the eye clinic for a diabetic eye exam without visual or ocular health complaints. His ocular history was pertinent for macular pigmentary disturbance, which was first noted by a previous care provider in 2001 and determined to be congenital. His systemic history was pertinent for:- Type 2 diabetes for approximately one year with a hemoglobin A1c of 6.7, controlled with diet and exercise.
- Hypertension, with the most recent blood pressure reading of 127/79, controlled with amlodipine 5mg, hydrochlorothiazide 25mg and atenolol 100mg, all once daily.
- Hyperlipidemia, with recent cholesterol of 197, HDL 40, LDL 132.8 and triglycerides 121, controlled with diet.
- Esophageal reflux, but the patient is currently asymptomatic and not being actively treated.
- Other systemic medications, including aspirin 81mg once daily, ibuprofen 600mg TID as needed and sildenafil 100mg, half a tablet as needed.
The patient had no known environmental or medical allergies. Entrance testing revealed the patient’s visual acuity was 20/20 OD and 20/20-2 OS, with pupils equal and reactive to light in both eyes without an afferent pupillary defect. Ocular motility was full and smooth in both eyes and confrontation visual fields were full in both eyes. Upon biomicroscopic examination, mild meibomian gland stasis was noted in both eyes with all other structures remaining normal. Intraocular pressure (IOP) was taken with applanation tonometry using 0.25% fluorescein sodium/0.4% benoxinate hydrochloride ophthalmic solution and was measured at 18mm Hg OD and 17mm Hg OS.
Dilation was performed with 2.5% phenylephrine hydrochloride ophthalmic solution and 1% tropicamide ophthalmic solution. The dilated exam revealed a 0.3 cup-to-disc ratio both horizontally and vertically with healthy rim tissue in both eyes. The macula was flat and remarkable for subtle pigment disruption, while the posterior pole was remarkable for scattered, well-defined hypopigmented/drusenoid-like formations in both eyes (Figure 1). The periphery was unremarkable in both eyes.
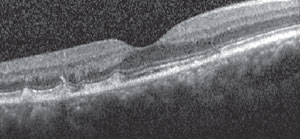 | |
| Fig. 2. The patient had basal lamina drusen-like formations at the level of the outer photoreceptor layer and anterior retinal pigment epithelium. |
Amsler grid testing was normal in the right eye, but revealed a small, less-than-one-degree area of metamorphopsia two degrees temporal to fixation in the left eye. Fundus photos documented the scattered hypopigmented lesions, and optical coherence tomography (OCT) yielded basal lamina drusen-like formations at the level of the outer photoreceptor segments and anterior retinal pigment epithelium (RPE) (Figure 2).
At this time, the differential diagnoses were: fundus flavimaculatus, multifocal pattern dystrophy and basal laminar drusen variant. The patient returned for fluorescein angiography (FA) and fundus autofluorescence (FAF) imaging to further refine the differentials. The FA showed hyperfluorescence of the lesions during early phases with normal choroidal flush and residual staining of the lesions in late phases (Figure 3). The autofluorescence imaging revealed scattered hyperfluorescent lesions in the posterior pole and hypofluorescence corresponding to the macular RPE disruption (Figure 4).
Subsequent to testing and consultation with a retina specialist, the patient was diagnosed with multifocal pattern dystrophy. This conclusion was based on the lack of progression in the fundus findings, FA findings and unaffected vision for more than 10 years from when the retinal findings were first documented. Treatment and management for the patient included regular monitoring of fundus with dilated fundus exam to rule out the formation of choroidal neovascularization, education about the inheritance pattern, and home Amsler grid testing.
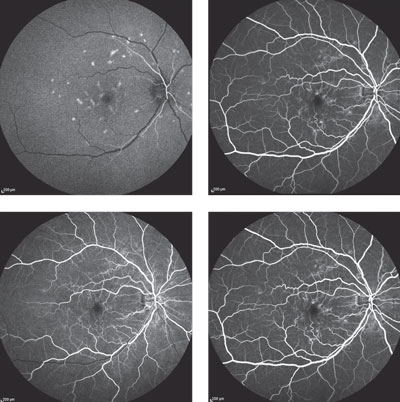 | 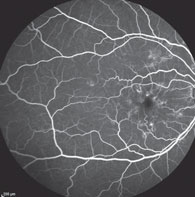 Fig. 3. Fluorescein angiography of the patient’s right eye at time stamps of 0:22, 0:26, 0:46, 1:10 and 3:27. Lesions are shown in the early phases with normal choroidal flush and residual lesion staining in the late phases. |
Discussion
Autosomal dominant pattern dystrophies can be broken into five groups:- Adult-onset foveomacular vitelliform pattern dystrophy.
- Butterfly-shaped pigment dystrophy.
- Reticular dystrophy of the RPE.
- Multifocal pattern dystrophy simulating fundus flavimaculatus.
- Coarse pigment mottling in the macula (fundus pulverulentus)
| Table 1. Five Types of Autosomal Dominant Pattern Dystrophies and Typical Characteristics | ||||
| Dystrophy | Typical Age of Onset | Typical VA | Retinal Presentation | |
| Adult-Onset Vitelliform Dystrophy | 4th-6th decades | 20/30-20/60 | Bilateral, circular, 1/3-1DD | |
| Butterfly-Shaped Pigment Dystrophy | 2nd-5th decades | 20/20-20/25 | Bilateral, triradiate hyperpigmentation | |
| Reticular Dystrophy | 5th decade | 20/30-20/70 | Bilateral, fishnet/ chicken wire hyperpigmentation pattern, 4-5DD | |
| Multifocal Pattern Dystrophy | 4th-6th decades | 20/20-20/40 | Bilateral, multiple yellow fleck-like lesions | |
| Fundus Pulverulentus | 4th-5th decades | 20/20-20/40 | Bilateral, coarse macular pigment mottling | |
Autosomal dominant pattern dystrophies are characterized by mild, midlife visual disturbances in the presence of various fundus findings.5,11 These visual disturbances typically progress very little, with the vision remaining mostly intact until late adulthood; however, a minority of patients will experience vision loss secondary to atrophy, choroidal neovascularization or both.1,8-10 While a majority of pattern dystrophies will present bilaterally, in some cases a different dystrophy will present in the fellow eye.
MPD is one of five types of autosomal dominant pattern dystrophies that have slight variations in vision, onset and retinal presentation (Table 1). These conditions are characterized by deep, intraretinal yellow, orange, or gray pigmentary deposits/changes.6,11,12 Pattern dystrophies have a variable rate of occurrence in the general population, as it can be inherited dominantly or can occur by various mutations in the peripherin/RDS gene; multifocal pattern dystrophy is inherited through autosomal dominant mutations in the peripherin/RDS gene on chromosome 6.3,11,13,14
While genotypically similar to other pattern dystrophies, multifocal pattern dystrophy is phenotypically unique. It can be distinguished from other autosomal dominant pattern dystrophies by its characteristic well demarcated, irregular, yellowish flecks, which take on a triradiate configuration.3,5,11 These flecks are scattered throughout the posterior pole and, in some cases, extend beyond the vascular arcades (Figure 5).3,5,11
Although multifocal pattern dystrophy will typically present with minimal to no visual effect and visual acuity from 20/20 to 20/40, a small area of central metamorphopsia may be seen with amsler grid testing, although this is more common in non-multifocal pattern dystrophy dystrophies.3-7,15
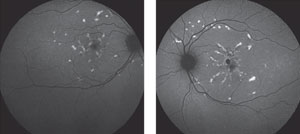 | |
| Fig. 4. Fundus autofluorescence images of the right and left eyes, respectively. The autofluorescence imaging revealed scattered hyperfluorescent lesions in the posterior pole and hypofluorescence corresponding to the macular RPE disruption. |
Differentials
Fundus flavimaculatus, Stargardt’s disease and a basal laminar drusen variant are three differentials to consider when diagnosing multifocal pattern dystrophy. Fundus flavimaculatus and Stargardt’s disease are arguably the most important differentials due to the similarity in retinal presentation.
Fundus flavimaculatus and Stargardt’s disease are variants of the same pathology, inherited autosomal recessively through the ABCA4 gene on chromosome 1.3,16,18,19 Both are characterized by irregular-shaped, triradiate, yellow, fleck-like lesions of variable size and shape within the posterior pole and concentrate around the retinal vascular arcades.3,18
While the two conditions are quite similar in terms of etiology and presentation, two important differences exist:
Onset. Stargardt’s disease typically develops early in life, while fundus flavimaculatus typically develops later in early to mid-adulthood, making the latter a more important differential as it will correlate more closely with age of onset for multifocal pattern dystrophy.18
Macular involvement. In Stargardt’s disease, the macula, paramacular region or both can have a central “beaten bronze” appearance.18 Patients afflicted with Stargardt’s disease will present with retinal abnormalities and experience vision loss much earlier in life, typically in the first or second decades.18 The vision loss associated with Stargardt’s disease is typically in the range of 20/100 to 20/200, or worse.16
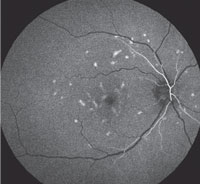 | |
| Fig. 5. Fluorescein angiography showing normal choroidal flush at 10 seconds during the early arteriole phase of the FA. |
Vision loss also occurs in fundus flavimaculatus, but occurs later in life and typically remains mild; it can decrease to 20/200, where it will typically stabilize.15
Stargardt’s and fundus flavimaculatus differ from multifocal pattern dystrophy in key ways:
Onset. While Stargardt’s disease typically presents in the first or second decades, multifocal pattern dystrophy presents in the fourth to sixth decades.
Effect on vision. Patients with multifocal pattern dystrophy typically maintain normal to mild reduction in acuity, whereas patients with Stargardt’s disease and fundus flavimaculatus see more severe visual impairment.
Fluorescein angiography. The diagnostic feature of Stargardt’s disease and fundus flavimaculatus is the dark choroid on FA due to the lipfuscin storage in the RPE cells.15,17,18,20,21 This does not happen in multifocal pattern dystrophy. Rather, a normal choroidal flush is present on FA (Figure 5).
The second differential to consider is a variant of basal laminar drusen. The retinal presentation of basal laminar drusen was first described in 1977 as small, raised, subretinal yellow lesions, and then further qualified in 1985 as focal areas of thickening in the RPE basement membrane.22-24 Around this same time, researchers qualified basal laminar drusen as cellular aggregations.22,25 These subretinal lesions can be widespread in the retina and mimic multifocal pattern dystrophy (Figures 6a and 6b).22
The best way to differentiate between multifocal pattern dystrophy and basal laminar drusen is through OCT, which will allows the clinician to qualify the type of deep retinal lesion by comparing images (Figures 6a and 6b).
Visual acuity also correlates between the two conditions, as basal laminar drusen may have minimal to no effect on vision; however, basal laminar drusen are also associated with pseudovitelliform macular detachments, which can result in subretinal fluid and mimic choroidal neovascular membranes.22,24 Basal laminar drusen formation can subsequently result in a progressive reduction in vision.22 Progressive vision loss is rarely seen in multifocal pattern dystrophy; choroidal neovascular membranes rarely occur.5,17
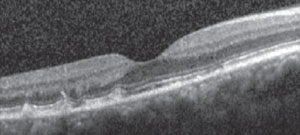 | |
| Fig. 6a. OCT image of our patient shows lesions at the level of outer photoreceptors and anterior retinal pigment epithelium. | |
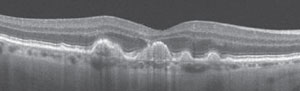 | |
| Fig. 6b. Optical coherence tomography of basal laminar drusen. Photo: eyewiki.aao.org. |
Treatment and Management
The treatment and management of multifocal pattern dystrophy centers around its etiology, visual effect and role as a risk factor for the development of choroidal neovascularization. In the rare case the patient develops choroidal neovascularization, the standard of care is a referral to a retina specialist for consideration of anti-vascular endothelial growth factor (VEGF) therapy.The key to managing the genetic component of the condition is patient education, which may entail a referral for genetic counseling. Through education, the patient and their family will be aware of the condition, its progression and prognosis.
Managing the visual effect of multifocal pattern dystrophy is as important as genetic counseling. Most cases of multifocal pattern dystrophy will retain normal to mildly reduced vision, in the 20/20 to 20/40 range; however, in rare instances vision loss can be more severe.3-7,15 When vision is not 20/20, it is important to manage its effect on the patient’s life. The clinician should address the patient’s ability to read a newspaper or book; read aisle signs at the grocery store; complete basic household repairs; and participate in hobbies such as playing cards, building models, etc.
The effect of vision loss on the patient’s life is best managed through low vision rehabilitation. Patients with multifocal pattern dystrophy can achieve the acuity needed for reading a newspaper or book and distance spotting through low magnification.
For reading, patients will likely benefit from single-vision reading glasses and may require monocular occlusion, depending on the add, acuity discrepancy and preferred retinal locus.
Monocular telescopes can be used for distance spotting, whether in the grocery store or walking down the street. For those patients who want to be able to take care of household repairs or continue with hobbies, such as building models, optivisors can improve visual performance. Optivisors can be used to provide an intermediate add while also providing lighting; a 10D loupe can increase magnification when needed. For close work coupled with intermediate vision, a pair of bifocal glasses can be used with an add in the distance portion for intermediate vision and an add in the bifocal portion for close work.
In addition to using conventional optical tints, which provide selective light filtering, task lighting can enhance contrast and reduce glare. Tinting is most successful with blue blocker tints (yellow, orange, amber), which reduce the glare experienced by patients due to the scattering of light on surfaces and by the ocular media.26-29 Tints can also improve vision quality and contrast sensitivity by decreasing light scattering and veiling glare to improve the retinal image.26-31
Conclusion
While genotypically similar to other pattern dystrophies, MPD is phenotypically unique with a close resemblance, funduscopically, to fundus flavimaculatus and Stargardt’s disease. The test of choice to aid in diagnosis is fluorescein angiography, which shows normal choroidal flush in multifocal pattern dystrophy and the pathognomonic dark choroid in fundus flavimaculatus and Stargardt’s disease. This delineation is key because of the better visual prognosis associated with multifocal pattern dystrophy.
A better prognosis is also found with multifocal pattern dystrophy when compared with basal laminar drusen. The test of choice in distinguishing these two conditions is OCT, because it allows for the identification of the different structures of the deposits and lesions. This differentiation becomes important because patients with basal laminar drusen are at an increased risk of developing a pseudovitelliform macular detachment, which can cause subretinal fluid that mimics choroidal neovascularization; in patients with multifocal pattern dystrophy, it is rare to have progressive vision loss or development of choroidal neovascularization.
Despite the better visual prognosis for multifocal pattern dystrophy over the two leading differentials, there is still the possibility of mild vision loss, since there is no current treatment for the condition. Management of the dystrophy involves patient education, referral to a retina specialist and treatment of a choroidal neovascular membrane, in the rare instance that it occurs. In the event of impaired vision, low vision rehabilitation to aid in activities of daily living is also critically important.
Dr. Hamilton, a residency-trained optometrist, is the director of Inpatient Low Vision Optometry and director of the Low Vision Optometry Externship Program at the Connecticut VA Healthcare System, West Haven.
Dr. Burke is a residency-trained low vision optometrist at the Connecticut VA Healthcare System.
1. Francis PJ, Schultz DQ, Gregory AM, et al. Genetic and phenotypic heterogeneity in pattern dystrophy. British Journal of Ophthalmology. 2005;89(9):1115-19.2. Souied EH, Rozet JM, Gerber s, et al. Two novel missense mutations in the peripherin/RDS gene in two unrelated French patients with autosomal dominant retinitis pigmentosa. Euro J Ophthal.1998;8(2):98-101.
3. Boon C, Van Schooneveld M, Den Hollander A, et al. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. British Journal of Ophthalmology. 2007;91(11):1504-11.
4. Fishman GA. Inherited macular dystrophies: a clinical overview. Australian and New Zealand Journal of Ophthalmology.1990;18(2):123-8.
5. Gass JD. Stereoscopic atlas of macular diseases: diagnosis and treatment. Vol 1. 4th ed. St Louis: CV Mosby, 1997:314-25.
6. Marmor MF, Byers B. Pattern dystrophy of the pigment epithelium. American Journal of Ophthalmology. 1977;84(1): 32-44.
7. Watzke RC, Folk JC, Lang RM. Pattern dystrophy of the retinal pigment epithelium. Ophthalmology. 1982;89(12):1400-6.
8. Zhang K, Garibaldi DC, Li Y, et al. Butterfly-shaped pattern dystrophy: a genetic, clinical, and histopathological report. Archives of Ophthalmology. 2002;120(4):485-90.
9. Marmor MF, Mcnamara JA. Pattern dystrophy of the retinal pigment epithelium and geographic atrophy of the macula. American Journal of Ophthalmology. 1996:122(3):382-92.
10. Yang Z, Lin W, Moshfeghi D, et al. A novel mutation in the RDS/peripherin gene causes adult-onset foveomacular dystrophy. American Journal of Ophthalmology. 2003;135(2):213-18.
11. Alkuraya H, Zhang K. Pattern Dystrophy of the Retinal Pigment Epithelium. Retinal Physician. 01 May 2010:9 May. 2013. www.retinalphysician.com/printarticle.aspx?articleID=104279.
12. Hsieh RC, Fine BS, Lyons JS. Pattern dystrophy of the retinal pigment epithelium. Archives of Ophthalmology.1977;95(3):429-35.
13. Fossarello M, Bertini C, Galantuomo MS, et al. Deletion in the peripherin/RDS gene in two unrelated Sardinian families with autosomal dominant butterfly-shaped macular dystrophy. Archives of Ophthalmology. 1996;114(4):448-56.
14. Nichols BE, Drack AV, Vandenburgh K, et al. A 2 base pair deletion in the RDS gene associated with butterfly-shaped pigment dystrophy of the fovea. Human Molecular Genetics. 1993;2(5):601-3.
15. Fu AD, Ai E, Mcdonald HR, et al. Hereditary Macular Dystrophies. Duane’s Clinical Ophthalmology. 2006 ed. Vol 3. Lippincott Williams and Wilkins.
16. Gerth C, Andrassi-Darida M, Bock M, et al. Phenotypes of 16 stargardt macular dystrophy/fundus flavimaculatus patients with known ABCA4 mutations and evaluation of genotype-phenotype correlation. Graefe’s Archives Clinical and Experimental Ophthalmology. 2002; 240(8):628-38.
17. Agarwal A. Heredodystrophic disorders affecting the pigment epithelium and retina. Gass’ Atlas of Macular Diseases. Vol. 1. 5th ed. Saunders;2012.
18. Kanski JJ. Fundus dystrophies. Clinical Ophthalmology: A Systematic Approach. 6th ed. New York: Butterworth Heinemann;2007.
19. Allikmets R. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature Genetics. 1997;17(1):122.
20. Ernest JT, Krill AE. Fluorescein studies in fundus flavimaculatus and drusen. American Journal of Ophthalmology. 1966;62(1):1-6.
21. Uliss AE, Moore AT, Bird AC. The dark choroid in posterior retinal dystrophies. Ophthalmology. 1987;94(11):1423-27.
22. Meyerle C, Smith T, Barbazetto I, et al. Autofluorescence of basal laminar drusen. Retina. 2007;27(8):1101-6.
23. Gass J. Stereoscopic atlas of macular disease: diagnosis and treatment. 2nd ed. St. Louis: Mosby, 1977.
24. Gass J, Jallow S, Davis B. Adult vitelliform macular detachment occurring in patients with basal laminar drusen. American Journal of Ophthalmology. 1985;99(4):445-59.
25. Russell S, Mullins R, Schneider B. Location, substructure and composition of basal laminar drusen compared with drusen associated with aging and age-related macular degeneration. American Journal of Ophthalmology. 2000;129(2):205-14.
26. Rosenblum YZ, Zak PP, Ostrovsky MA, et al. Clinical research note: spectral filters in low-vision correction. Ophthalmic and Physiological Optics. 2000;20(4):335-41.
27. Leat JS, North RV, Bryson H. Do long wavelength pass filters improve low vision performance?. Ophthalmic Physiological Optics. 1990;10(3):219-24.
28. Zigman S. Vision enhancement using a short wavelength light-absorbing filter. Optometry and Vision Science. 1990;67(2):100-4.
29. Zigman S. Light filters to improve vision. Optometry and vision Science. 1992;69(4):325-28.
30. Faye EE. Clinical Low Vision. Boston: Little Brown & Co.;1984.
31. Longhurst RS. Geometric and Physical Optics. London: Longman; 1970.
