
|
A 22-year-old white male presented with a chief complaint of decreased vision, which had progressively worsened during the past two years.
Several previous eye care providers informed the patient that he needed to see a retina specialist. Because of insurance-related complications, however, he never sought specialized retinal care.
The patient had no documented ocular history. He appeared to be in good systemic condition, and said that he didn’t take any medications. Further, he reported no known allergies of any kind.
|
|
 |
|
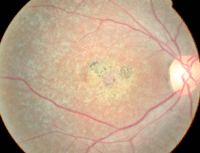
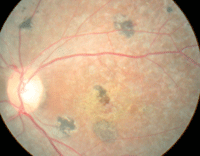
| This 22-year-old male presented with decreased vision in both eyes (OD top, OS bottom). What is the correct diagnosis? |
Diagnostic Data
Best-corrected visual acuity measured 20/80 OU at distance and 20/50 OU at near. External examination uncovered some central distortion on facial Amsler testing, which was confirmed on grid assessment.
We documented no evidence of afferent pupillary defect OU, but red color interpretation was unequal between the two eyes. There was no brightness desaturation. Refraction uncovered no changes OU.
The anterior segment evaluation was normal in both eyes. Intraocular pressure measured 16mm Hg OU. The pertinent posterior segment findings are illustrated in the photographs.
Your Diagnosis
How would you approach this case? Does this patient require any additional tests? What is your diagnosis?
Discussion
Additional testing might include a D-15 color vision test, fundus photography and a formal visual field test to evaluate for functional deficits. Sodium fluorescein angiography would not be indicated, unless choroidal neovascularization (CNV) was suspected.
The diagnosis is Stargardt’s disease––a congenital retinal dystrophy that is categorized on the continuum of macular degenerative diseases.1-7 In 1909, Carl Stargardt first described the condition as a “flecked retina disease” that caused decreased visual acuity in the first or second decades of life.1 Today, many clinicians continue to refer to Stargardt’s disease as “juvenile macular degeneration.”1-4
Optometrist Larry J. Alexander calculates that the incidence of Stargardt’s Disease in the US is one in 8,000 to 10,000 individuals.3
Characteristically, the condition presents in four distinct fundus patterns:3,4
- Macular degeneration without flecks
- Macular degeneration with perifoveal flecks
- Macular degeneration with diffuse flecks
- Diffuse flecks without macular degeneration
Stargardt’s has an autosomal recessive transmission pattern, which typically yields symmetric, bilateral presentations.3,5 The hallmark symptom is diminished central visual acuity; other classic signs include myopic refractive error, mild photophobia, glare disability, central scotomata and worsening acuity with accompanying color vision deficits in later disease stages.5,6
While the onset of symptoms usually occurs in the first or second decades of life, many patients remain asymptomatic until the fourth or fifth decades.1-6 In the early stages of disease, Stargardt’s patients exhibit a “bulls eye” maculopathy on the fluorescein angiogram. Additionally, the retinal pigment epithelium (RPE) develops a “beaten bronze” sheen which is apparent to clinical observers as the lesion progresses.3 Choroidal neovascularization has been documented as a late-stage complication.3
The pathophysiology of Stargardt’s includes three primary stages:1
- A defective rim protein (a glycoprotein associated with the rim of the photoreceptor outer-segment), encoded by the ABCA4 gene, causes an accumulation of protonated N-retinylidene-PE in the rod outer segments.
- Next A2E, a toxic byproduct of N-retinylidene-PE, accumulates in the RPE cells and causes tissue damage.
- The photoreceptors eventually succumb secondary to the ongoing compromised RPE function.
Because RPE destruction results in photoreceptor loss, progressively worsening visual consequences are inevitable.1-7
Even in the 21st
century there is no known method of halting the process.
Clinicians should discuss the benefits of genetic counseling with patients at risk for Stargardt’s disease. This will help affected individuals better understand the disease, the risk of congenital transmission and the overall likelihood of complete vision loss.
Those diagnosed with Stargardt’s disease can benefit from low vision rehabilitation, psychological counseling and work-related counseling.2,3,6 A functional knowledge of ocular genetics can help in the diagnosis of the condition, as well as understanding the potential for other associated syndromes.3 Finally, because the disease is capable of producing symptoms without signs in young patients, when the visual acuity is reduced––whether the patient is aware of it or not––in the setting of an eye with no other visual pathology or amblyogenic vectors, the clinician should consider testing for Stargardt’s disease before simply laying off the diagnosis as amblyopia.1-5
We sent the patient to the retinology department to confirm our suspicion. Once testing was completed, we referred the patient to a low vision specialist for education on the most suitable optical and non-optical aids.
We will continue to see the patient for routine eye care, and will refer him to retinology for annual testing and monitoring.
2. Bither PP, Berns LA. Stargardt's disease: a review of the literature. J Am Optom Assoc. 1988;59(2):106-11.
3. Alexander LJ. Herditary retinal-choroidal dystrophies. In: Alexander LJ. Primary Care of The Posterior Segment. Norwalk, Conn.:Appleton and Lange; 1994:425-77.
4. Wakabayashi K, Yonemura D, Kawasaki K. Electrophysiological analysis of Stargardt's disease fundus flavimaculatus group. Doc Ophthalmol. 1985;60(2):141-7.
5. Noble KG, Carr RE. Stargardt's disease and fundus flavimaculatus. Arch Ophthalmol 1979; 97(7):1281-5.
6. Miedziak AI, Perski T, Andrews PP, et al. Stargardt's macular dystrophy--a patient's perspective. Optometry. 2000;71(3):165-76.
7. Lachapelle P, Little JM, Roy MS. The electroretinogram in Stargardt's disease and fundus flavimaculatus. Doc Ophthalmol. 1989;73(4):395-404.
