 Step right up, ladies and gentlemen, and buy some Hamlin’s Wizard Oil! It’s the “best pain remedy on Earth.” As advertised, “There is no sore it will not heal! No pain it will not subdue!” Among its many benefits, it cures rheumatism, sore throat, headache, toothache, backache, as well as cramps, colic, diarrhea and cancer. (Never mind that it’s made of 50 to 70% alcohol as well as camphor, ammonia, chloroform, sassafras, cloves and turpentine.)
Step right up, ladies and gentlemen, and buy some Hamlin’s Wizard Oil! It’s the “best pain remedy on Earth.” As advertised, “There is no sore it will not heal! No pain it will not subdue!” Among its many benefits, it cures rheumatism, sore throat, headache, toothache, backache, as well as cramps, colic, diarrhea and cancer. (Never mind that it’s made of 50 to 70% alcohol as well as camphor, ammonia, chloroform, sassafras, cloves and turpentine.)
About a century ago, Hamlin’s Wizard Oil was one of the most popular patent medicines—and possibly one of the least harmful—of its kind. Back then, there were no requirements that a remedy prove its safety and efficacy, or even reveal its ingredients.
While our current drug approval process is by no means perfect, the US Food and Drug Administration has arguably some of the world’s most rigorous procedures for determining whether a drug is safe, consistent and effective. Nowadays, we take for granted that the many pharmaceuticals we use provide a positive effect without causing harm (if taken as directed). We prescribe drugs every day, yet rarely do we consider how the drug came into existence. This article provides an inside look at this laborious but necessary process.
The Drug Approval Process
Here’s a breakdown of how a drug undergoes the FDA’s approval process, which takes an average of eight to 12 years. Bear in mind that the FDA itself does not test drugs; its role is to review test data and to approve or disapprove a drug to be marketed for a specific medical use.
• Preclinical studies. The process begins with the identification of a unique molecule that is believed to have promise in treating human disease. Preclinical testing involves toxicology, pharmacokinetics and dosing studies conducted on animals (often rodents and dogs) to help establish dosing limits and potential adverse effects. It is critical that potential drug adverse effects are identified prior to the commencement of human testing. (See “The FDA Drug Approval Process,” opposite.)
• Investigational application. After obtaining promising data from preclinical studies, the pharmaceutical company must first file an Investigational New Drug Application (IND) in order to begin the clinical testing on humans. The IND document is a roadmap of the proposed study. It includes the study protocol, the relevant data from the pre-clinical studies, drug manufacturing data and, most importantly, the informed consent document and other data relevant to the recruitment of human subjects. The FDA has 30 days to review and approve the IND application.
• Phase I studies. The Phase I study examines the pharmacologic actions and safe dosage range of a drug. Two numbers are very important: Therapeutic Index (TI) and No Observable Adverse Effect Level (NOAEL). The TI is the ratio of the lethal dose in 50% of test subjects (LD50) to the effective dose in 50% of test subjects (ED50). Any ratio below 20 is considered dangerous. Because lethal doses are never used in human studies, the NOAEL is a useful analog to the TI for human trials.
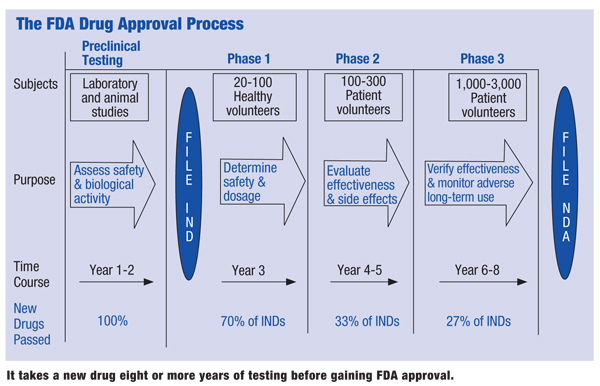 |
| It takes a new drug eight or more years of testing before gaining FDA approval.
|
• Phase II studies. Phase II studies not only help identify unsafe medications and drug side effects, but also begin to identify drug efficacy. For this reason, the selected test subjects have the condition for which the drug is intended. This means Phase II studies are true safety/efficacy trials, so this particular study can make or break a drug. By their nature, Phase II studies evaluate a small group of individuals. If efficacy and safety cannot be established at this level, then it is impossible for the drug study to proceed with a larger population of test subjects.
A critical component of a Phase II study is the establishment of a treatment dose. This includes both the amount of drug and its proper dosing interval. Dose the patient too low and the drug demonstrates no efficacy; too high and it produces unacceptable toxic side effects. One technique to establish a “best dose” is to randomize test subjects into low-, medium- and high-dose groups. This is a more efficient method of quickly identifying dosages that are unacceptably low or high. All Phase II studies are double blind, with the control group receiving a placebo dosage.
• Phase III. If Phase II studies demonstrate drug efficacy and safety, then this most expansive phase of drug testing is justified. Phase III studies involve a large number of patients who have the disease that the study drug intends to control or cure. A critical element that differentiates Phase III from Phase II studies is the dosage form. The test dose in Phase III must be in the form that is intended to be approved and marketed to the public. So, dosing information from Phase II trials is critical in designing the final dosage form.
Next in importance is the protocol of the study. Proper patient selection is critical. Investigators must establish inclusion and exclusion criteria, and carefully define the drug indication.
The goals of therapy (“endpoints”) must also be well delineated. Test populations and control groups need to be large enough to yield statistical relevance, and the groups must be matched by age, gender and other pertinent factors to ensure statistical credibility.
To improve statistical credibility in certain drug trials, multiple, parallel Phase III studies are performed. Additionally, an extension of a Phase III study may occur, with subjects continuing drug therapy under “open label” conditions, where the actual treated subjects are identified.
In addition to safety and efficacy, the Phase III studies must demonstrate, via animal studies, acceptable levels of the drug’s mutagenic potential, or the tendency to induce cancer in patients who receive the drug.
At the formal completion of the Phase III trial, the data are evaluated and the safety and efficacy profile of the drug is established, as well as the initial recommended dosage amount and frequency. Drug side effects, warnings and contraindications are also established. Most importantly, the initial drug indication is identified.
• NDA. At the conclusion of Phase III studies (assuming these prove successful), the pharmaceutical company files a New Drug Application (NDA) with the FDA. All study information—including efficacy and safety information from drug trials, manufacturing data and pharmaceutical data—must be included in this extensive document.
• Phase IV studies. Following FDA approval, Phase IV studies (called post-approval or post-marketing studies) have several purposes. First, their design must mirror that of the other preclinical studies, although the rigor of design is less intense. A Phase IV study can be used to expand the indications for a drug that was approved for limited applications, or it can be a comparator study (a study intended to demonstrate clinical superiority over a competitor’s product).
 Generic Drugs
Generic Drugs
A drug patent in the US lasts 20 years, and during that time other manufacturers cannot produce the same agent. But once a drug has reached the end of its 20-year patented life, it can be produced and marketed by other companies, under certain circumstances, in a generic form. Just as for new, branded drugs, the FDA is also responsible for the approval of generic (or “copycat”) forms of approved pharmaceutical agents.
The Drug Price Competition and Patent Term Restoration Act of 1984—more commonly known as the Hatch-Waxman Act—was designed to promote the availability of generic drugs.
It accomplished this by increasing financial incentives for new drug development and by abbreviating the process of generic drug approval.
The law allows generics to obtain FDA approval by submitting bioequivalence studies (as opposed to the more costly clinical data required to approve a totally new drug), and it extends the marketing exclusivity of branded drugs by up to five years (which is in addition to the 20-year patent duration of the original molecule). This extension compensates the developer for the additional time and money required to bring a new therapeutic agent to market.
A controversial and often abused component of this law allows the original pharmaceutical company to have an automatic stay of 30 months to challenge the generic manufacturer’s right to copy the branded drug. This is commonly used to stall the generic company’s ability to sell a cheaper version of the drug and maintain exclusivity over the sales of the drug.
Generic Drug Equivalence
The FDA will approve a generic version of a drug following the filing of an Abbreviated New Drug Application (ANDA) if the generic version is determined to be both therapeutically and pharmaceutically equivalent. It must also demonstrate bioequivalence. The specific meanings of these terms are critical to the use of a generic agent that delivers the same therapeutic benefits as the original, branded agent.
• Pharmaceutical equivalence. This implies that the generic contains the same active ingredients, the same dosage form and strength, and the same route of administration as the original drug. According to the FDA, generic ophthalmic solutions are required to have the same active and inactive ingredients in the same concentrations.
• Bioequivalence. This term means that the drug follows the same pharmacokinetic profile as the original—its absorption, distribution, metabolism and excretion can be expected to be the same as the reference drug. This implies that the drug reaches the same blood levels and half-life (bioavailability) of the original drug.
However, topical (locally-applied) agents—including ophthalmic, otic (ear) and parenteral (injectable) solutions—do not have to meet this requirement. Generic manufacturers can request a waiver of bioequivalence for topical products.
The Hatch-Waxman Act provides an incentive to generic drug manufacturers to be the first to file an ANDA. The first company to file a successful ANDA is allowed to exclusively market the new generic drug for 180 days. This advantage has led to a new class of generic drug—the “authorized” generic. This is a practice whereby the original drug manufacturer agrees to license the drug to a specific generic manufacturer, thereby retaining some control of the drug and profits derived from the sale of the generic form.
 Drug Withdrawals and Recalls
Drug Withdrawals and Recalls
Believe it or not, once the FDA approves a drug, it cannot withdraw it from the market. The FDA must negotiate with the manufacturer to voluntarily remove the pharmaceutical agent in question.
One of the most recent and important withdrawal actions was against the blockbuster NSAID Vioxx (rofecoxib, Merck). After Vioxx was approved and became widely used for its analgesic and anti-inflammatory benefits, the incidence of cardiovascular complications dramatically increased in patients who used it. At the request of the FDA, Merck withdrew the drug.
The sister drug of Vioxx, Celebrex (celecoxib, Pfizer), was allowed to stay on the market only after its manufacturer dramatically altered the package insert with a “black box” warning.
This warning requires the patient to be counseled about a drug’s serious side effects and requires that the physician obtain the patient’s informed consent to use the drug.
Patients do not select their own treatments. They depend upon and trust that their physicians will prescribe drug therapies that are both safe and effective; that is, drug treatments that have the potential to produce greater benefits than risks. So, it’s important that clinicians understand how the FDA functions to protect patients from medications that can produce significant harm. It is equally important that prescribers understand FDA reporting mechanisms to bring adverse drug effects to the FDA’s attention. n
Dr. Onofrey is a clinical professor and the executive director of CE programs at the University of Houston College of Optometry, and a frequent lecturer on ocular disease management and pharmaceutical agents.
