The frequency of retinal detachments (RD) is estimated to be approximately one per 10,000 people per year.1 The most common risk factors include myopia (especially high myopia), aphakia, trauma, retinal detachment in the fellow eye and a family history of retinal detachment. In addition, a variety of peripheral retinal abnormalities are associated with an increased risk of a retinal detachment.
While you cannot lower a person’s absolute risk of developing a retinal detachment, by prioritizing patient education on RD signs and symptoms, your patients will know to call your office as soon as symptoms present. Seeing these patients early is imperative, as early intervention has a better chance of limiting visual loss from RD.
Optometrists must manage and educate their patients who have asymptomatic retinal breaks or peripheral retinal lesions that are known to be associated with a higher risk of developing RD. They must also take care of referral recommendations for patients with symptomatic or otherwise high-risk horseshoe retinal tears.
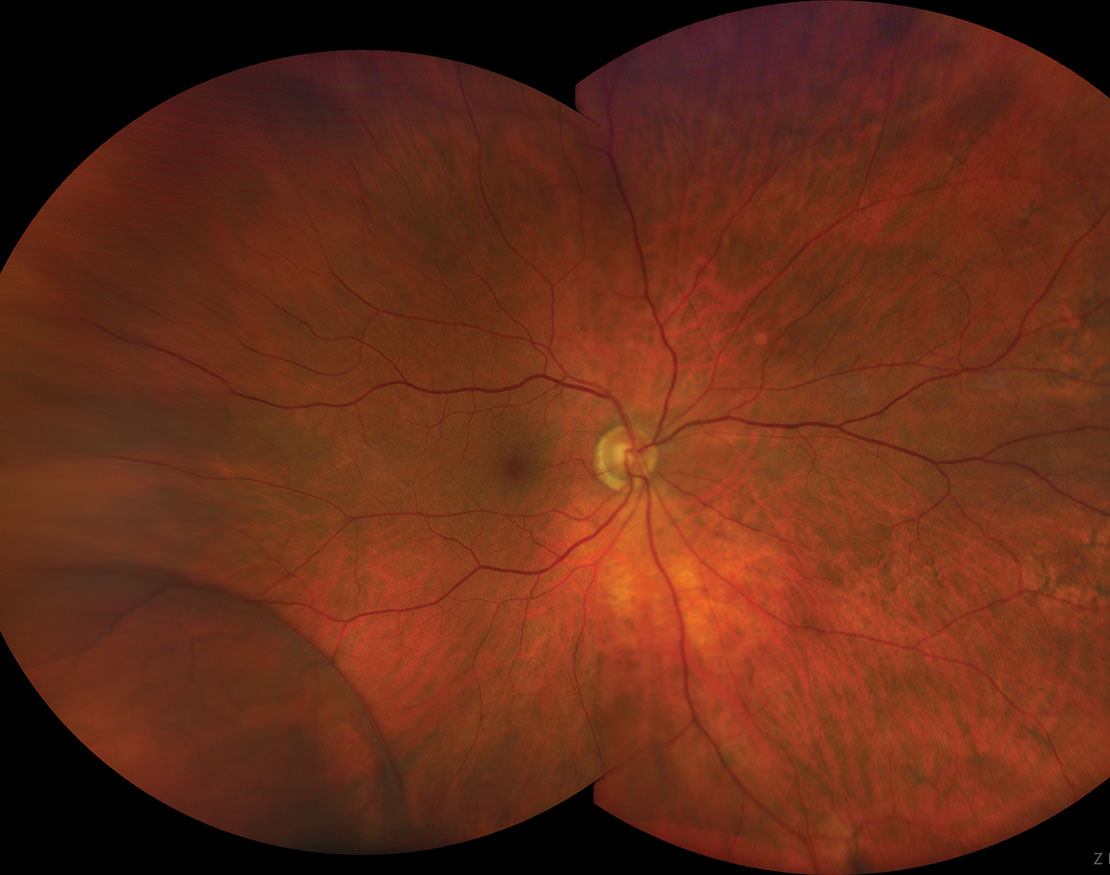 |
| In most cases, a retinoschsis will neither impact vision nor carry any symptoms. However, on rare occasions, it can evolve into a retinal detachment. Click image to enlarge. |
Posterior Vitreous Detachment
A posterior vitreous detachment (PVD), which can lead to a full-thickness retinal break, is the most common event that precedes RD development. A PVD occurs when the vitreous separates from the surface of the retina and collapses anteriorly toward the vitreous base. The vitreous base is a broad adherence of the vitreous to the peripheral retina near the ora serrata. The well-known symptoms of a PVD are flashes and floaters.
Vitreous traction on the peripheral retina causes these flashing lights, or photopsia, in the patient’s vision. If RD is present, patients may also present with what they might describe as a curtain, shade or a shadow obscuring their side vision. Ask all patients at risk for RD whether they see these signs or if they feel like they have lost a portion of their peripheral vision.
Patients who present with a symptomatic PVD without peripheral retinal breaks require no immediate treatment; however, it is pertinent to re-examine these patients within one to two weeks, as retinal breaks may develop days to weeks after the onset of symptoms.
When examining a patient with an acute PVD, carefully look at the vitreous cavity and note the presence or absence of pigment cells (a tobacco dust sign or Shafer’s sign) or the presence of red blood cells consistent with a vitreous hemorrhage. In 10% to 15% of patients, a retinal tear is a sign of an acute PVD.2,3 If the symptomatic patient has red blood cells or a vitreous cavity hemorrhage, there is a 70% likelihood of an associated retinal tear.4
Patients who present with asymptomatic PVD and have peripheral retinal degeneration at a higher risk of developing a retinal tear. Warn these patients of the signs and symptoms and to return immediately if these symptoms occur.
If a patient returns and is symptomatic, you should perform a dilated examination with scleral depression in addition to widefield photography, if available. Be sure to document what drops used to dilate the patient, the lens you used to examine the fundus (90D, 20D, 28D, etc.) and whether you performed scleral depression.
Lattice Degeneration
This is present in 7% to 8% of the general population, and, of those cases, up to 45% are bilateral.5 Patients with lattice lesions are at a 1% risk of developing RD in their lifetime.1 Lattice degeneration is a lesion characterized by sharp demarcated margins, oval or round in appearance and may vary in pigmentation but have common white lines in the crossing retinal vessels.
Another characteristic of lattice degeneration is a pocket of liquefied vitreous anterior to the lesion as well as firm vitreoretinal attachments on the margins. These adherent vitreoretinal attachments are the reason that this lesion carries a risk of retinal tear and detachment following an acute PVD. In a large review of clinical cases, 30% of eyes who suffered an acute RD had lattice degeneration, and, in 83% of these cases, the associated retinal tear was within the bed of the lattice lesion.6
Prophylactic treatment of lattice degeneration is not indicated; however, patients with photopsia or increased floaters (symptomatic) should be followed closely or referred for a retinal evaluation.
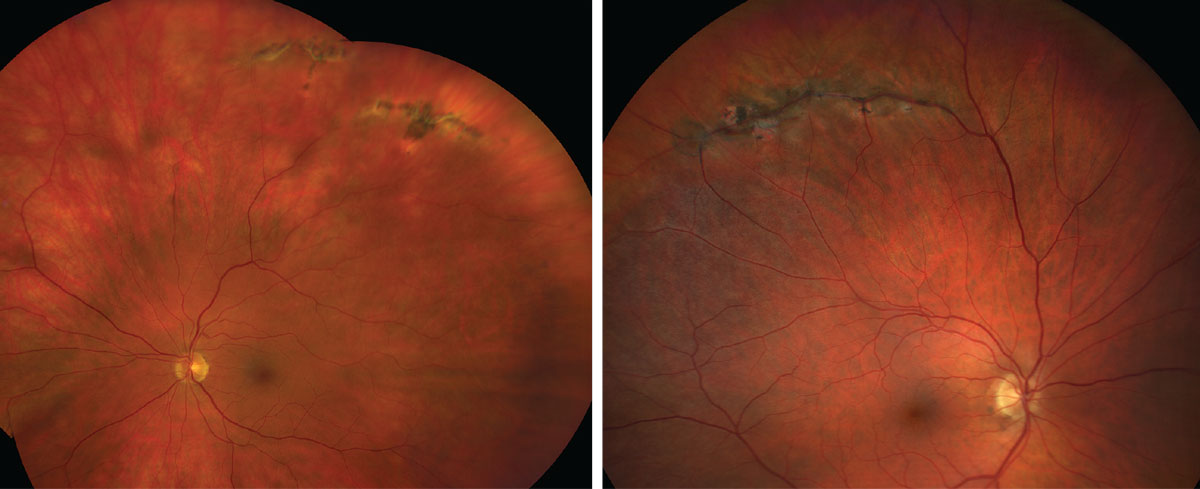 |
| Lattice degeneration, as seen here, is associated with an increased risk of a retinal detachment. Click images to enlarge. |
Cystic Retinal Tufts
These are noted in approximately 5% of the population and are thought to be a congenital abnormality in the development of the peripheral retina.7 Peripheral retinal degenerations are found during a dilated examination with indirect ophthalmoscopy or documented with widefield retinal imaging of the peripheral fundus. In appearance, a cystic retinal tuft is chalky white in color, round or oval in shape and primarily composed of glial tissue.
Vitreous condensations are attached to the surface and base of the cystic retinal tuft and may be associated with pigmentary changes secondary to the chronic vitreous adhesion. These small anomalies in the peripheral fundus may be hard to appreciate without use of scleral depression.
The incidence of developing RD from a cystic retinal tuft is approximately 0.28%.8 There is no recommendation for treatment of the cystic retinal tuft alone.
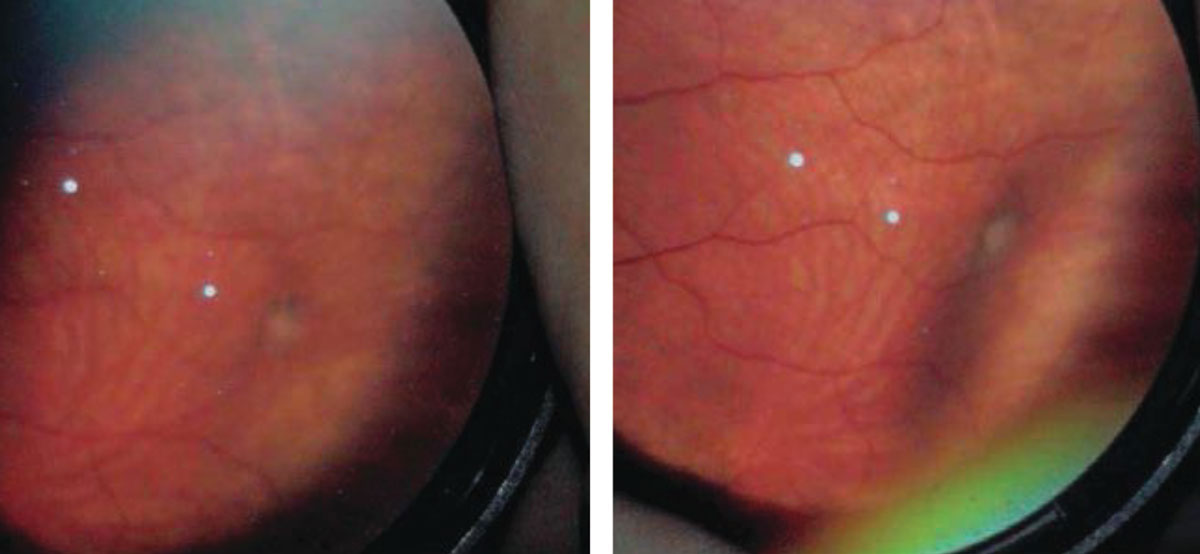
Retinoschisis
Acquired degenerative retinoschisis is thought to be idiopathic and present in 1% of the population, with up to 82% of cases being bilateral.9 Although retinoschisis is typically a benign peripheral lesion, vision-threatening complications may occur over time. Retinoschisis is most commonly seen in the inferotemporal quadrant, although it can occur in the superior temporal and superior nasal quadrants as well. The clinical features of a retinoschisis include a smooth, dome-like appearance that is transparent and allows visualization of the RPE and choroid. It is immobile, with an abrupt transition between the schisis cavity and normal posterior retinal anatomy.
The natural history of a retinoschisis reveals that it rarely progresses more posterior from where it first appeared, and the incidence of RD from a retinoschisis is less than 2.5%.10 Although the incidence of RD arising from progression of a retinoschisis is low, the mechanism is associated with the formation of an inner wall hole and an outer wall break, which is defined as a full-thickness retinal defect.
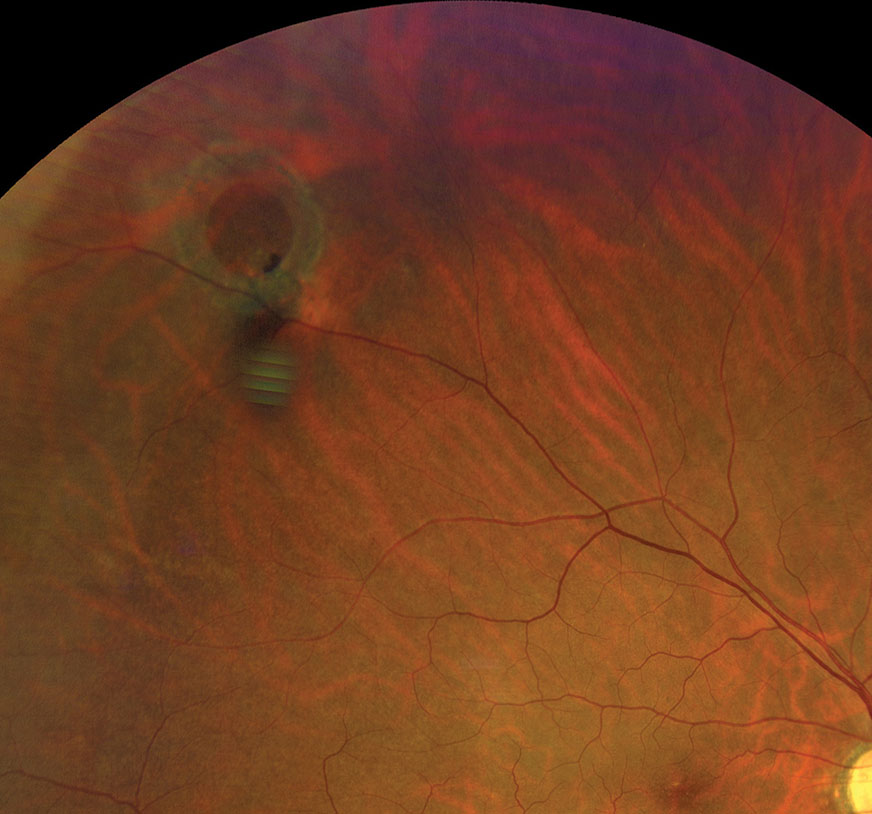 |
| Refer operculated retinal hole patients with symptoms associated with RD to a retina specialist. Click image to enlarge. |
The appearance of an inner wall hole is small and round, resembling an atrophic retinal hole, whereas outer wall retinal breaks are usually much larger and located more near the posterior margin of the retinoschisis. Outer wall breaks occur in approximately 17% of retinoschisis whereas inner wall holes are much less frequently at less than 4%.10
Although the incidence of RD secondary to retinoschisis is low, using optical coherence tomography can be very helpful to rule out RD if the schisis lesion appears to be migrating posterior.
Given the low incidence of an associated RD, there is no formal recommendation for prophylactic treatment. I tell these patients that in most cases a retinoschsis will not impact vision nor carry any symptoms, but, on rare occasions, it can evolve into RD. Discuss the signs and symptoms of RD with the patient and perform a dilated examination annually.
Atrophic Retinal Holes
The underlying cause of these defects is a poorly functioning choriocapillaris that no longer gives adequate circulation to the retinal layers above the hole. This chronic thinning and decompensation of the chorioretinal blood supply has a domino-like effect on the overlying neurosensory retina, resulting in thinning.
Atrophic retinal holes are present in approximately 5% of the overall population and often identified on routine dilated examination with or without widefield imaging. When present in conjunction with lattice degeneration, the incidence of atrophic holes is higher. Atrophic holes may be found in isolation, within areas of lattice degeneration (18%) or adjacent to lattice degeneration (42%).11
The incidence of RD from an atrophic retinal hole is exceptionally low, as atrophic holes are not a result of vitreous traction. The etiology of an atrophic retinal hole is secondary to a focal degeneration of the neurosensory retina from abnormal blood supply to the retinal pigment epithelium from the underlying choriocapillaris. It is vital to perform a careful examination with scleral depression and determine if there is any associated subretinal fluid surrounding the hole. Use annual dilated exams or serial widefield retinal imaging to observe asymptomatic atrophic retinal holes.
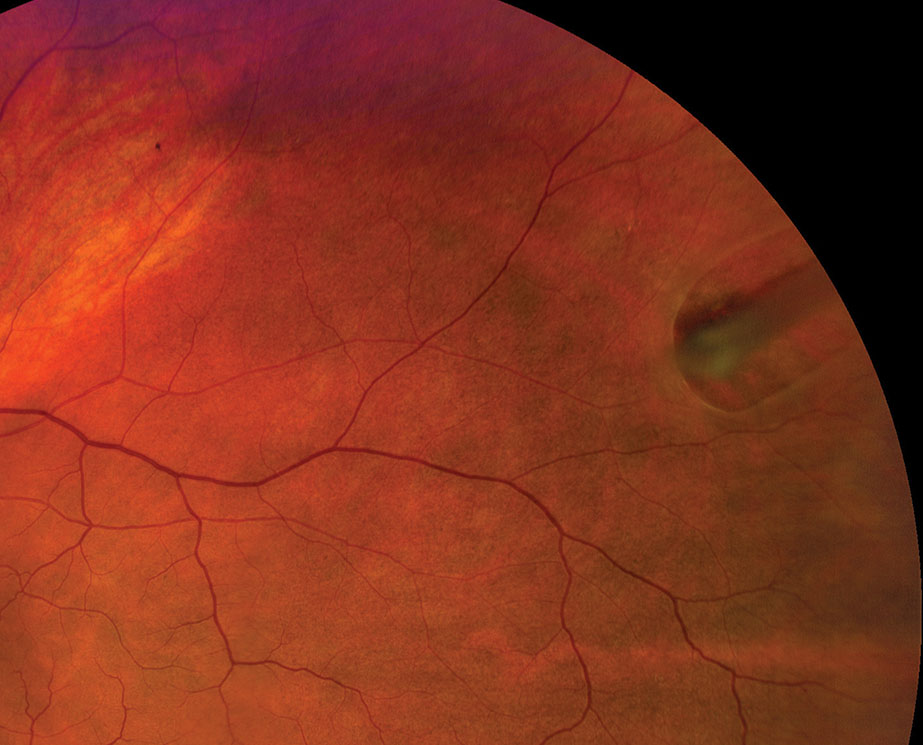 |
| Refer any horseshoe retinal tear case emergently for prophylactic treatment. Click image to enlarge. |
Operculated Retinal Holes
Unlike atrophic retinal holes, operculated retinal holes originate secondary to vitreous traction and may carry a higher incidence of RD if they fail to seal spontaneously. The operculum signifies a release of vitreous traction, and, in most cases, the edges of the hole will flatten and seal spontaneously. If a patient presents with acute symptoms and an operculated retinal hole, evaluate with scleral depression and determine if there is any residual vitreous traction on the anterior surface of the retina or subretinal fluid.
One clinical pearl based on my clinical experience is, when the size of the operculum is five times smaller than the associated retinal hole, this lesion has reached end stage.
If the patient has operculated retinal holes as well as associated symptoms of flashing lights and floaters, then refer to a retinal specialist.
Horseshoe Retinal Tears
Horseshoe retinal tears are the cause of most detachments. The estimated incidence of a horseshoe retinal tear in a patient with an acute PVD is 8%.12 Acute PVD, in which there is a strong adhesion of the vitreous to the retinal surface, causes the horseshoe retinal tear. Although most vitreous will spontaneously separate from the retinal surface during a PVD, areas where there is a firm vitreoretinal adhesion traction can result in tearing the retina and cause a full thickness defect. These retinal breaks are associated with a higher incidence of RD, as liquefied vitreous will traverse through the retinal break and accumulate in the subretinal space separating the neurosensory retina from the retinal pigment epithelium.
Any horseshoe retinal tear, whether symptomatic or asymptomatic, should be referred emergently for prophylactic treatment to prevent progression to RD.
Prophylactic Treatment
Consider prophylactic retinopexy for symptomatic peripheral retinal lesions as well as horseshoe retinal tears, even for those without symptoms of flashing lights or floaters, given the high risk for progression to a retinal detachment. Prophylactic treatment can include laser photocoagulation or cryotherapy. The decision on which method to use is typically based on the location of the pathology, as more anterior lesions require cryotherapy. The end result of prophylactic treatment is a chorioretinal reaction around the lesion that prevents the accumulation or spread of subretinal fluid.
The optometrist’s role in post-treatment care depends on the treating retina specialist. The optometrist’s responsibilities may include widefield retinal imaging of the treated lesion, dilated examination with scleral depression as well as ongoing education of symptoms of RD.
For any patient with peripheral retinal pathology associated with a risk of RD, it is important to educate them on the signs and symptoms and follow-up accordingly. When patients present with symptoms of RD, dilate them and perform BIO with scleral depression to rule out a retinal tear or detachment. If you are not comfortable managing symptomatic patients, make a referral to a specialist for a second opinion.
Evaluating these retinal defects is not enough, as the complications may escalate and cause significant vision loss. Preventing the effects of retinal detachment requires proper management and decisive action to implement effective management strategies. More importantly, clinicians must communicate with their patients about the risk of retinal detachment associated with peripheral retinal pathology.
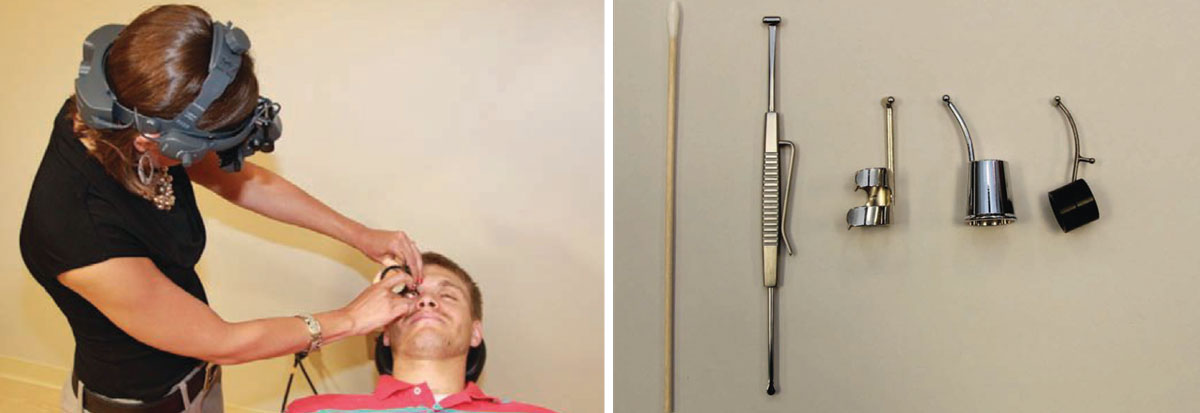
How to Perform Scleral DepressionScleral depression combined with binocular indirect ophthalmoscopy (BIO) lets you evaluate the retinal periphery, which may not be well viewed with slit-lamp biomicroscopy or BIO alone. Scleral depression indents the retinal periphery to bring it into better view with BIO. I typically perform scleral depression on areas that BIO indicates further evaluation, such as area suspicious for peripheral retinal pathology such as lattice degeneration, atrophic retinal holes, cystic retinal tufts or retinoschisis.
There are two techniques for performing scleral depression: transdermal (i.e., through the skin) and transconjunctival, in which you place the scleral depressor directly on the bulbar conjunctiva. I use the transdermal technique more often because it is more comfortable for the patient. A topical anesthetic is not necessary when the transdermal technique is performed correctly. The transconjunctival technique is necessary for examining the temporal and nasal aspects of the peripheral fundus or when you attempt to depress a posterior lesion. This technique requires a topical anesthetic such as tetracaine, and the scleral depressor should be placed posterior to the ciliary body. When performed correctly, transconjunctival scleral depression should cause only minimal discomfort for the patient. Whether with the transdermal or transconjunctival technique, be sure not to apply pressure on the lacrimal gland. To examine the superior retina, ask the patient to look down, then place the scleral depressor at the base of the lashes. Next, instruct the patient to look superiorly as you concomitantly move the depressor along the contour of the globe. The objective of scleral depression is to view the far retinal periphery and evaluate peripheral retinal lesions. By gently indenting the globe, you can also study the effects of motion and contrast on various lesions. Scleral depression also allows you to see whether a hole or tear is open or sealed by pigmentation, which can help you decide whether to refer a patient to a retina specialist for treatment. Scleral depression is necessary for patients who have symptoms or a history of retinal disease because it enables you to see the entire retina, to the ora serrata, to observe or rule out a change in retinal pathology. Perform scleral depression in the opposite quadrant or location of patient symptoms, as superior pathologies produce inferior symptoms. Do not perform scleral depression on patients who have had recent blunt ocular trauma, as it could exacerbate a penetrating injury, or on patients who have recently undergone surgery, including cataract and LASIK procedures, because you could open the eyeball or dislodge the flap. Also avoid scleral depression in patients who have angioid streaks unless it is necessary; it could induce perforation of the globe in such cases. When documenting your findings of the peripheral retinal examination, note what techniques and lenses you used.
|
Dr. Haynie is an adjunct clinical profressor at Pacific University College of Optometry and practices at Sound Retina in Tacoma, WA. He is a fellow of the American Academy of Optometry and a member of the Optometric Retina Society.
1. Wilkes SR, Beard CM, Kurland LT, et al.. The incidence of retinal detachment in Rochester, Minnesota, 1970-1978. Am J Ophthalmol. 1982;94:670-3. 2. Linder B. Acute posterior vitreous detachment and its retinal complications. Acta Ophthalmol. 1966;Suppl 87:1-108. 3. Tasman WS. Posterior vitreous detachment and peripheral retinal breaks. Trans Am Acad Ophthalmol Otolaryngol. 1968;72:217-24. 4. Benson WE. Retinal detachment: diagnosis and management. 2nd ed. Philadelphia: Harper & Row Publishers Inc, 1988: 4. 5. Foos R, Simons K. Vitreous in lattice degeneration. Ophthalmology 1984;91:452-7. 6. Dumas J. Schepens CL. Chorioretinal lesions predisposing retinal breaks. Am J Ophthalmol. 1966;61:620-30. 7. Straatsma BR, Foos RY, Feman SS. Degenerative disease of the peripheral retina. In Duane DD, ed. Clinical Ophthalmology, Volume 3, Chapter 26. Philadelphia, Harper & Row, 1986:1. 8. Byer NE. Cystic retinal tufts and their relationship to retinal detachment. Arch Ophthalmol. 1981;99:1788. 9. Byer NE. Clinical study of senile retinoschisis. Arch Ophthalmol. 1968;79:36. 10. Byer NE. Long-term natural history study of senile retinoschisis with implications for management. Ophthalmology. 1986;93:1127-37. 11. Tasman WS. Peripheral Retinal Lesions. In: Yanoff M, ed.Ophthalmology. 2nd ed. Philadelphia: Mosby; 2004. 12. Westfall AC, Maa A, Mieler W, Holz E. Incidence of retinal tears and late-onset retinal breaks in eyes with symptomatic posterior vitreous detachment. Invest Ophthalmol Vis Sci. 2004;45:2066. |
